
毛细管柱气相色谱法测定水产品中三氯杀螨醇残留量
2018-08-31 10:39:30吴文慧
江西化工 2018年4期
吴文慧 李 亮
(1.江西省食品药品安全监控中心,江西 南昌 330029; 2.宜春市食品稽查支队,江西 宜春 336000)
三氯杀螨醇(dicofol)化学名为:2,2,2-三氯-1,1-双(4-氯苯基)乙醇,是一种有机氯杀螨剂。可防治棉花、果树、蔬菜等农作物上的螨类,对成螨、若螨、卵有很强的触杀和胃毒作用,药效期和速效期长。研究表明,该药物对人畜有一定毒性,主要中毒症状为头痛、头晕、多汗、胸闷、瞳孔散大、视物不清,以及恶心、呕吐、腹泻,局部接触可引起接触性皮炎。
水产品中三氯杀螨醇检测的研究较少,关于植物源性食品中该农药的检测报道较多,主要采用气相色谱-电子捕获检测法[1]-[3]、气相色谱-质谱法[4]。为了更好应对国外技术壁垒,建立相应的检测方法是非常必要的。
本文利用丙酮-水对样品进行提取,石油醚液液分配去除脂类杂质,磺化除去样品中的油脂,碱化将三氯杀螨醇转化为邻苯二甲酸二丁酯(dibutyl phthalate,DBP)后,用配有电子捕获检测器的气相色谱仪测定,外标法定量,建立了毛细管柱气相色谱-电子捕获法测定水产品中三氯杀螨醇残留量检测方法。
1 实验部分
1.1 仪器与试剂
气相色谱(Agilent 7890,Agilent公司),配微电子捕获检测器;旋涡混合器(IKA MS2,德国);离心机(上海安亭)TDL-5-A;旋转浓缩仪(瑞士BUCHI)R-215;氮吹浓缩仪(美国Organomation)NEVP-24;有机系针孔滤膜(天津津腾)0.22μm。
1.2 试剂和药品
正己烷,丙酮均为色谱纯;无水乙醇、石油醚、浓硫酸、氢氧化钾为分析纯;提取溶剂:丙酮:石油醚=1:1(体积比);10mol/L氢氧化钾溶液;20g/L硫酸钠溶液;配制溶液用水为超纯水;三氯杀螨醇标准物质(Dr.E公司):纯度≥99.0%。
2 方法和结果
2.1 前处理步骤
2.1.1 提取
称取(5.0±0.01g)试样置于50mL塑料离心管中,加入5mL水、3g氯化钠,混匀,加入15mL提取溶剂,涡旋2min,4000r/min离心5min,转移上清液于150mL的鸡心瓶中。对固体残渣依照上法重复提取两次,合并提取液。
2.1.2 磺化
将提取液于40℃旋转蒸发至近干,加入10mL正己烷复溶残渣。将正己烷复溶液转移至25mL玻璃管中,加入5mL浓硫酸,轻摇30s,3000r/min离心5min,取出正己烷层于另一25mL干净玻璃管中。下层剩余溶液中再加入3mL新鲜正己烷涡旋,3000r/min离心5min,合并正己烷层。
2.1.3 碱化
加入4mL 10mol/L氢氧化钾溶液、0.5mL的无水乙醇,充分涡旋混合2min以碱化完全。静置1min,待分层后弃除下层水相。加入2×5mL 20g/L硫酸钠溶液洗涤有机相。将正己烷在氮吹仪上于40℃浓缩至干。加入1mL正己烷充分溶解残渣,过滤膜,供气相色谱-电子捕获检测器分析。
2.2 标准溶液的制备
标准储备液(100μg/mL):准确称取三氯杀螨醇标准品0.0050g,置于50mL容量瓶中,用正己烷溶解并定容至刻度,摇匀,-4℃冰箱内避光保存;标准中间液(10μg/mL):在室温下取标准储备液1mL置于10mL容量瓶中,用正己烷稀释定容,4℃冰箱内避光保存。
2.3 GC-μECD分析条件
色谱柱:石英毛细管柱DB-XLB 60m×0.25mm×0.25μm,或相当者;载气和尾吹:氮气(纯度≥99.999%),载气流量:1.0mL/min,尾吹流量:60.0mL/min;色谱柱温度:初始温度为60℃保持1.0min,以25℃/min升温至270℃,保持8.0min;进样口温度:240℃;检测器温度:300℃;进样方式:不分流进样;进样量:1μL。
2.4 线性范围和定量限
在本方法所确定的实验条件下,在0mg/L~1.0mg/L范围内呈良好的线性关系,其线性方程:
Y=1.079e2X+5.494e2,相关系数为0.9999。
当添加水平为0.01mg/kg时,信噪比(S/N)大于10,可满足定量检测要求,方法的定量限可达0.01mg/kg,能够满足我国对水产品中三氯杀螨醇监控的需求。
2.5 回收率和重复性
本方法采用向阴性鲫鱼、草鱼样品中添加的方式进行方法学评估。试验时分别添加0.01mg/kg、0.02mg/kg、0.04mg/kg三个水平浓度进行回收率测定,每个水平进行6次平行测定,回收率范围在74.2%~110.2%之间,相对标准偏差均小于12.6%,具体结果见表1所示,能够满足农兽药残留检测的需求。
3 讨论
3.1 提取溶剂的选择
三氯杀螨醇能溶于大多数脂肪族和芳香族有机溶剂。参考文献[5],选用丙酮-石油醚(V/V =1:1)混合液作为提取溶剂,分三次提取,以便达到最好的提取效果;加入水以便更好的分散样品,适量氯化钠可以起到盐析作用,使样品中的待测物残留更容易被提取出来;石油醚同水相之间进行液液分配还避免了一些水溶性杂质被带入提取液中,减少了杂质的干扰。
3.2 净化方式的选择
提取液中仍有大量类脂物等杂质,如不经进一步处理,对检测结果和仪器必会造成影响甚至破坏。研究人员起初采用凝胶渗透色谱(GPC)进行净化,通过标准添加的方式确定了采集时间段、杂质丢弃时间等条件。但在进行方法学验证时发现添加回收率非常不稳定,在40%到90%之间波动较大。通过仔细排查,发现标准溶液、添加样品的色谱行为均出现了异常情况,在原来的保留时间附近出现了另一个峰,该峰的响应与标准溶液浓度同样呈线性关系,但与三氯杀螨醇的峰加合则不呈线性。经质谱分析,标准溶液所出两个峰之一为三氯杀螨醇,而另一个是邻苯二甲酸二丁酯(DBP)。
经进一步的资料查找,研究人员了解到,发现有相关[6]-[7]报道表明,在碱性或高温条件下,三氯杀螨醇容易分解成DBP,分解途径见图1所示,但分解率不确定,难以瞬间定量分解。而气相色谱仪进样口衬管的玻璃毛带弱碱性、检测器的温度比较高等因素都可能使三氯杀螨醇转化为DBP,这样的话,简单地检测三氯杀螨醇在定性和定量方面均难以控制。
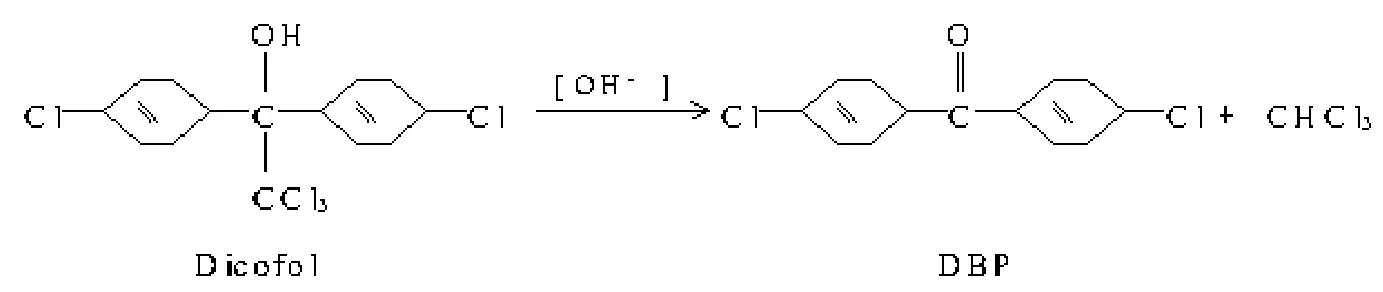
图2 三氯杀螨醇转化为DBP的途径
参考了部分文献[8]方法后,研究人员决定将三氯杀螨醇碱解后测定其碱解产物DBP。为了避免本底中可能含有DBP而对测定产生干扰,决定先磺化再碱解。磺化采用的浓硫酸不仅起到除去DBP的作用,还可以除去油脂类杂质;之后再用10mol/L KOH碱化,再用20g/L硫酸钠溶液洗涤,以达到碱解转化和进一步除杂的效果。
4 结论
本方法适用于鲫鱼、草鱼等水产品中痕量三氯杀螨醇残留量的测定,有效避免了三氯杀螨醇在前处理和色谱分析过程中降解的异常,方法的灵敏度、重复性等指标可以满足我国对三氯杀螨醇的监控的需求。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15 11:42:26
当代水产(2021年10期)2022-01-12 06:20:40
应用化工(2021年4期)2021-05-20 09:43:36
化工管理(2020年26期)2020-10-09 10:05:16
佳木斯大学学报(自然科学版)(2020年1期)2020-02-28 05:30:52
山东化工(2019年2期)2019-02-21 09:29:32
中成药(2017年12期)2018-01-19 02:06:26
食品与机械(2017年5期)2017-07-05 13:24:36
农产品加工(2017年6期)2017-05-09 18:04:52
环境科技(2016年4期)2016-11-08 12:18:58
