
FBP1基因突变致果糖1,6二磷酸酶缺乏的癫持续状态1例并文献复习
2018-08-03 05:03张赟健
中国循证儿科杂志 2018年3期
张赟健 路 通 王 艺
1 病例资料

既往史:患儿出生后第2 d出现呕吐伴低血糖、高胆红素血症,于外院治疗,病程中无抽搐,无呼吸窘迫。患儿平时食用含果糖食物如苹果、梨和西瓜等后易出现恶心、呕吐、精神差、乏力。
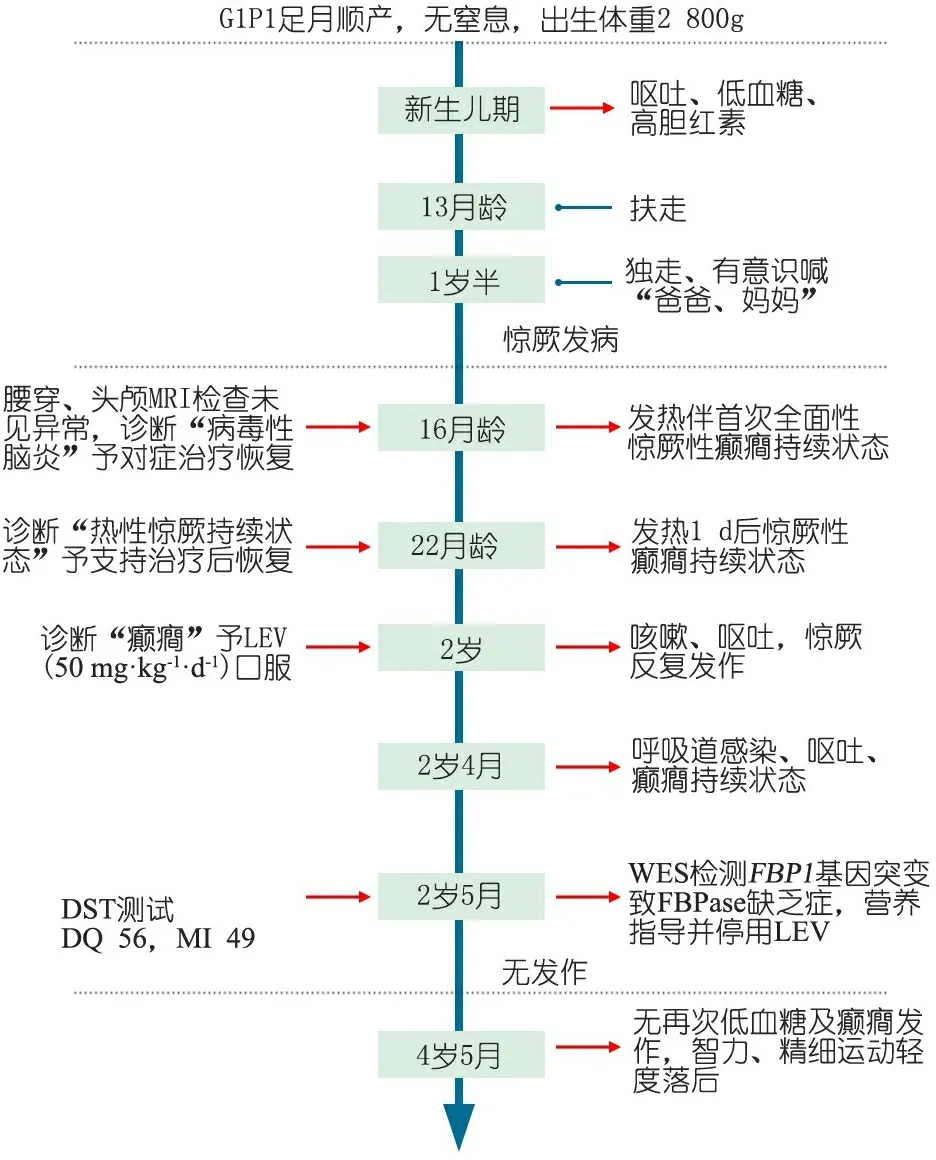
图1 临床信息时间轴
注 LEV:左乙拉西坦;WES:全外显子测序
入院查体:神志清,精神反应好,皮肤未见异常色素斑,无特殊面容,颅神经检查未见异常,眼球活动好,双侧瞳孔等大等圆,对光反应灵敏,伸舌居中,颈软,心肺听诊未见异常,腹软,无压痛,肝肋下可扪及2~3 cm,质软,脾肋下未触及,四肢肌力和肌张力正常,腹壁反射、腱反射可引出,克氏征、布氏征(-),病理反射(-),无震颤,共济运动未见异常。
辅助检查:血糖4.1 mmol·L-1,血乳酸0.9 mmol·L-1,ALT 43 U·L-1,AST 48 U·L-1,血气分析pH 7.4,血及尿质谱分析未见明显异常。腹部B超示肝轻度肿大。发作间期清醒与睡眠EEG检查均未见异常。颅脑MRI检查未见异常。
获得患儿父母知情同意后,对患儿家系行全外显子检测(WES)并行Sanger验证,结果显示患儿存在FBP1基因(NM_000507)复合杂合突变(exon5: c.704het_delC和exon7: c.959het_dupG)。Sanger测序对患儿父母进行验证,父亲携带c.704het_delC突变,母亲携带c.959het_dupG突变(图2)。FBP1基因突变可导致果糖1,6二磷酸酶(FBPase)缺乏症,是一种罕见的遗传代谢病,为常染色体隐性遗传。
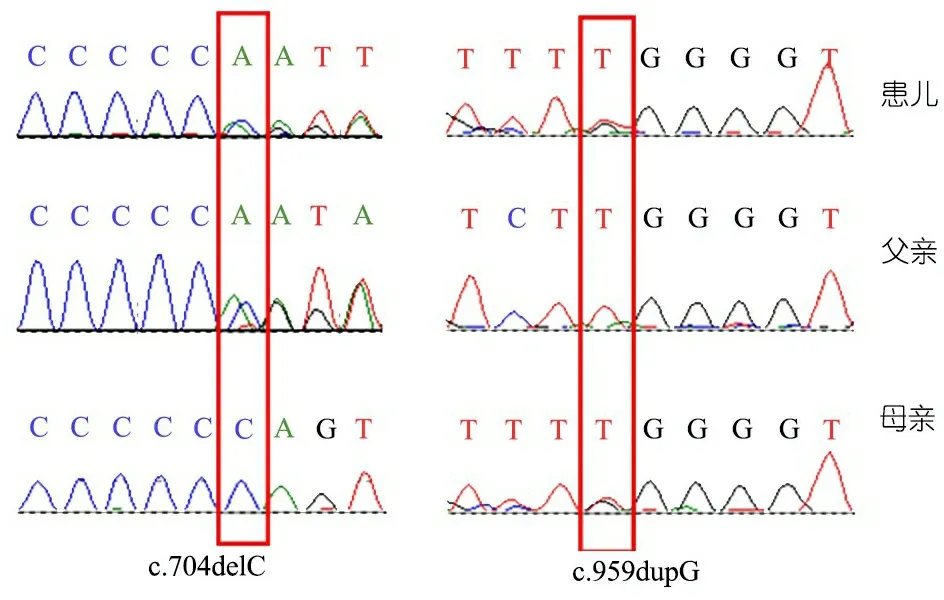
图2 患儿及其父母FBP1基因测序图
根据患儿致病基因突变位点情况向家属解释FBP1基因突变所致FBPase缺乏症的临床特征和遗传模式等基因诊断和产前诊断的相关问题。患儿母亲再次怀孕胎儿携带复合杂合突变从而致病的概率为1/4,于孕期行羊水穿刺及胎儿基因检测,胎儿携带母亲来源的FBP1:c.959het_dupG突变,而另1条染色体未携带父源的FBP1:c.704het_delC突变,为杂合突变携带者,理论预测不会出现FBPase缺乏症的临床表现。
2 讨论
FBPase由位于染色体9q22.2-q22.3的FBP1基因编码,El-Maghrabi等[5]1995年报告了FBP1基因结构和染色体定位,其由7个外显子组成。自Kikawa等[6]于1995年首次报道FBP1基因突变以来,全球在FBPase缺乏症患者中共已发现38种FBP1突变类型[7, 8]。图3显示,其中10种突变位于7号外显子。图4显示,多数患者突变位于7、5和1号外显子。7号外显子中c.959dupG为最常见的突变类型。Kikawa等对11个日本家庭的13例FBP1基因突变患者进行分析,c.959dupG突变率46%[9]。
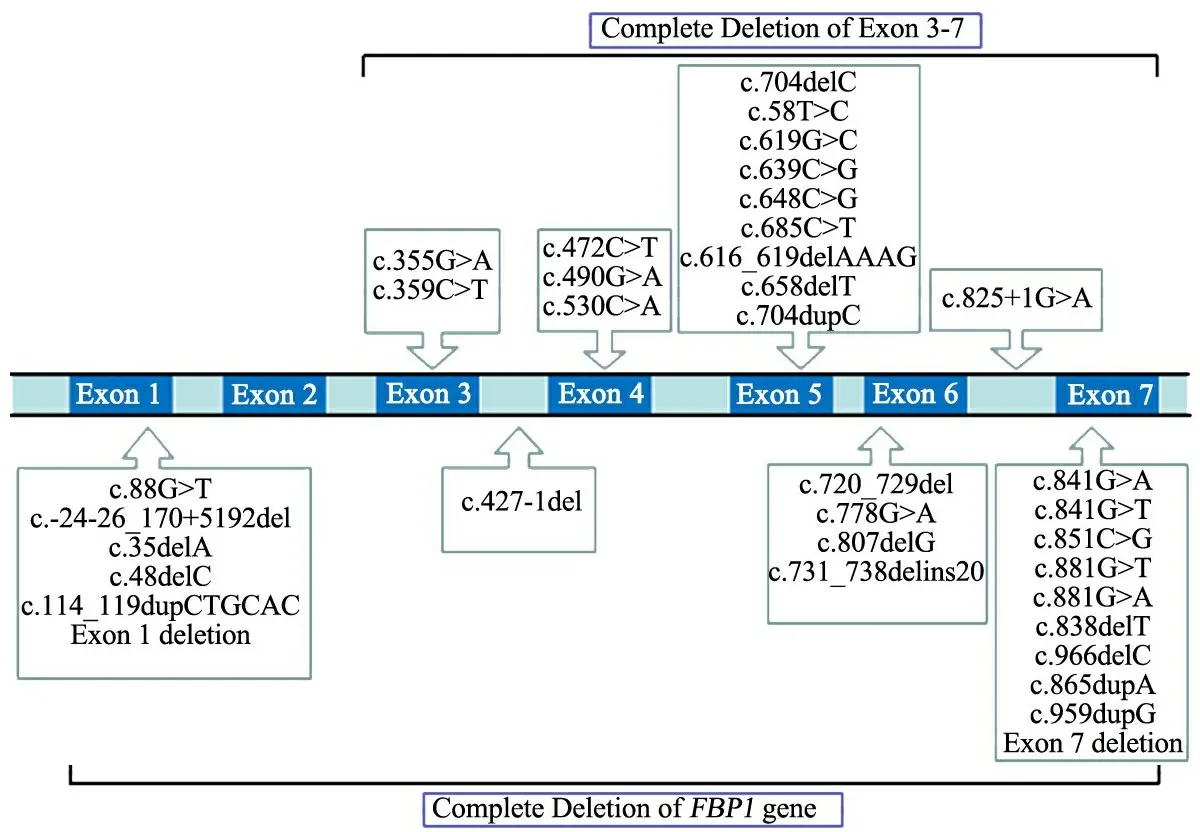
图3 FBPase缺乏症致病性FBP1基因突变位点图
本文患儿也携带该位点突变,且该位点的纯合突变病例也见于国内报道[10];欧洲和北美患者的该突变比例约为14%[4, 11]。因此,该位点为日本与中国患者人群的常见突变位点。另1常见突变位点为在巴基斯坦及阿拉伯人群中报道的c.841G>A[12]。Faiyaz-Ul-Haque等[13]在近亲婚配的阿拉伯患者中发现6个核苷酸的重复插入突变c114_119dupCTGCAC和c.841G>T无义突变。c.685C>T突变在摩洛哥患者中较常见[4]。此外,在日本、韩国及中国患者中检测到c.490G>A突变[7, 9, 14]。本文患儿和另1例中国患儿[7]存在c.704delC突变。其他位点突变尚未发现有重复报道。FBP1基因不同的突变类型表明该病存在显著的遗传异质性。c.959dupG、c.490G>A和c.704delC可能是东亚人群的主要突变位点。
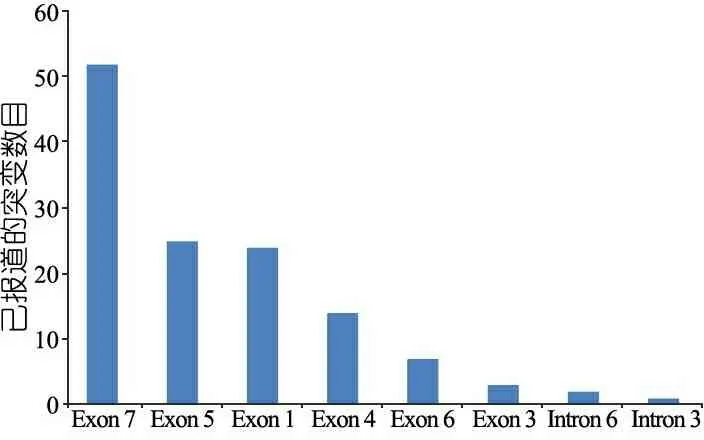
图4 已报道FBP1基因不同外显子/内含子携带突变的等位基因数
表1 73例FBPase缺乏症患者的临床和实验室检查信息

病例数[n(%)]性别 男34 (46.6) 女39 (53.4)起病年龄 新生儿期23 (31.5) 婴儿期27 (37.0) 1~3岁13 (17.8) >3岁10 (13.7)近亲婚配18 (24.7)诱发因素 摄入少36 (49.3) 感染16 (21.9) 其他因素或不明21 (28.8)临床表现 消化道症状32 (43.8) 意识障碍25 (34.2) 呼吸窘迫25 (34.2) 肝肿大20 (27.4) 惊厥17 (23.3) 发育迟缓6 (8.2)实验室检查 低血糖69 (94.5) 代谢性酸中毒68 (93.2) 高乳酸血症46 (63.0) 高尿酸血症18 (24.7) 酮症16 (21.9) 肝功能异常16 (21.9)总计73 (100)
90%以上的FBPase缺乏症患儿存在低血糖和代谢性酸中毒,大多数有高乳酸血症,但这些生化指标变化并无特异性。患儿同时存在酮尿表明糖异生途径受到影响(图5)[22]。此外,低血糖和酸中毒患儿尿中检测出甘油,尤其是甘油-3-磷酸,可为FBPase缺乏症的诊断提供重要线索[19]。早期病例通过肝活检进行肝组织FBPase酶活性测定可明确诊断[22]。随着基因检测及分子诊断技术的发展,目前可通过FBP1基因突变检测确诊。对于临床高度疑似患儿,若未发现FBP1基因突变,则需肝活检行FBPase酶活性测定[23]。本文患儿通过WES发现FBP1基因复合杂合突变,最终确诊为FBPase缺乏症。
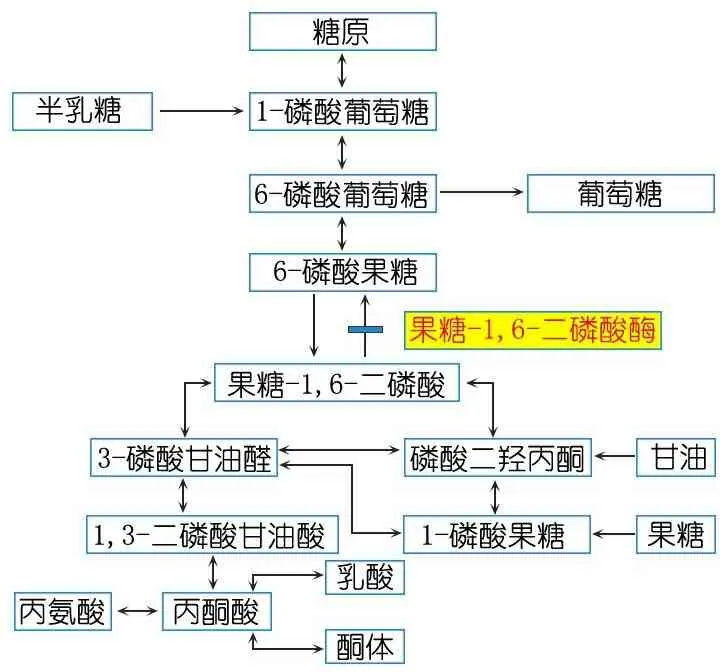
图5 糖异生途径示意图
治疗FBPase缺乏症的主要原则是维持正常的血糖水平。维持治疗期主要是避免长期禁食、饥饿和感染,增加喂养次数,限制高蛋白、高脂肪食物的摄入,避免过多摄入果糖、蔗糖或山梨糖醇[17]。发热或精神差时立即予葡萄糖口服或静脉注射。急性感染期必要时可使用胃管持续喂养以保持稳定的血糖浓度。Hasegawa等[20]报告,甘油果糖治疗可发生脑水肿等严重的神经系统并发症或致命危险。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
当代医药论丛(2022年17期)2022-10-09
河南畜牧兽医(2022年2期)2022-01-01
今日农业(2021年16期)2021-11-26
种子(2021年3期)2021-04-12
中国生殖健康(2020年4期)2021-01-18
大自然探索(2020年5期)2020-06-19
中国生殖健康(2018年4期)2018-11-06
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
