
近红外光谱技术快速测定附子及其炮制品中双酯型生物碱含量*
2018-07-31 08:51杨学军
中国药业 2018年14期
邓 芳,杨学军,罗 准
(湖南省衡阳市食品药品检验检测中心,湖南 衡阳 421000)
附子为毛茛科植物乌头 Aconitum carmichaelii Debx.子根的加工品,为回阳救逆之要药,有补火助阳、散寒止痛功效。因其含有毒性较大的乌头碱、新乌头碱及次乌头碱双酯型生物碱,2015年版《中国药典(一部)》[1]规定了双酯型生物碱的上限量。按《中国药典》采用高效液相色谱(HPLC)法测定含量,检验周期长,效率低,且对照品价格昂贵,试剂消耗量大,成本高。本试验中通过采集附子及其炮制品的近红外光谱,采用一阶导数加矢量归一法快速测定附子及其炮制品中的毒性成分,进行快捷、准确地初筛,从而提高检验效率。现报道如下。
1 仪器与试药
1.1 仪器
MPA近红外光谱仪(德国布鲁克公司);Agilent 1260系列高效液相色谱仪(美国Agilent公司);Mettler AE240型电子分析天平(瑞士梅特勒公司);TTL-DCⅡ型氮吹仪(北京同泰联科技发展有限公司);DK410HT型超声仪(深圳市得康洗净电器有限公司);泰斯特F80W型高速万能粉碎机(天津市泰斯特仪器有限公司)。
1.2 试药
乌头碱对照品(批号为110720-201312,纯度为98.9%),新乌头碱对照品(批号为110799-201307,纯度为 98.3%),次乌头碱对照品(批号为 110798-201308,纯度为98.6%),均购自中国食品药品检定研究院;乙腈、四氢呋喃为色谱纯;水为超纯水;其他试剂均为分析纯。50批附子及炮制品样品购自四川江油和陕西[2]2个主产区,经我中心傅黎春主任中药师鉴定为毛茛科植物乌头 Aconitum carmichaelii Debx.的子根与加工品。
2 方法与结果
2.1 HPLC法测定含量
色谱条件:色谱柱为Agilent XDB-C18柱(250 mm×4.6mm,5μm,编号为 990967-902);检测波长为 235nm;柱温为 30℃;进样量为10 μL;流速为1.00 mL/min;流动相以乙腈-四氢呋喃(25∶15)为流动相 A,以0.1 mol/L醋酸铵溶液(每1 000 mL加冰醋酸0.5 mL)为流动相B,按表1条件进行梯度洗脱。

表1 流动相梯度洗脱条件
溶液制备:称取新乌头碱对照品0.009 46 g、次乌头碱对照品0.009 96 g、乌头碱对照品0.011 30 g,精密称定,分别置100 mL容量瓶中,加异丙醇-二氯甲烷(1∶1)混合溶液 40 mL,超声处理(功率 300 W,频率40 kHz,水温在25℃以下)使溶解,定容至刻度,作为贮备液。精密量取贮备液5 mL,置100 mL容量瓶中,加异丙醇-二氯甲烷(1∶1)混合溶液并定容至刻度,摇匀,即得混合对照品溶液。取本品粉末(过3号筛)约2 g,精密称定,置具塞锥形瓶中,加氨试液3 mL,精密加入异丙醇-乙酸乙酯(1∶1)混合溶液50 mL,称定质量,超声处理(功率300 W,频率40 kHz,水温在25℃以下)30 min,放冷,再称定质量,用异丙醇-乙酸乙酯(1∶1)混合溶液补足减失的质量,摇匀,滤过,精密量取续滤液25 mL,40℃以下减压回收溶剂至干,残渣精密加入异丙醇-二氯甲烷(1∶1)混合溶液3 mL溶解,滤过,取续滤液,即得供试品溶液。
系统适用性试验:理论板数以新乌头碱计为42 700,分离度大于1.5。重复进样5次,计算含量的 RSD,结果新乌头碱为1.2%,次乌头碱为1.3%,乌头碱为1.2%(n=5)。
样品含量测定:分别精密吸取对照品溶液与供试品溶液各10 μL,注入液相色谱仪,测定。色谱图见图1。

图1 混合对照品溶液高效液相色谱图
结果计算:根据测得的峰面积,按外标法计算供试品中双酯型生物碱含量,以新乌头碱(C33H45NO11)、次乌头碱(C33H45NO10)和乌头碱(C34H47NO11)的总量计。结果见表2。
2.2 样品的NIR光谱采集
将收集的43批附子及炮制品,粉碎,过3号筛[3-4],每份5 g,分装入样品瓶中,混合均匀。光谱采样条件:环境温度25℃,相对湿度45% ~70%,积分球漫反射,分辨率8cm-1,光谱扫描范围4000~10000cm-1。原始光谱图见图2。
图2 43批附子及炮制品的近红外光谱图
2.3 定量模型的建立和优化
光谱预处理方法选择[5-10]:为消除噪声和基线漂移对测定结果可靠性与准确性的影响,对收集到的近红外光谱进行预处理。以模型的交叉检验决定系数(R2)和校正均方根差(RMSEC)作为参数来评价模型,通过比较,最终确定一阶导数光谱+矢量归一化法对光谱进行预处理。预处理方法对RMSEC和 R2的影响见表3。
谱段选择:手动优选最佳波段,辅以交叉验证均方差(RMSECV)作为模型性能的评价指标,选取常用的偏最小二乘法(PLS)进行数据回归分析。在6 101.8~4 597.6 cm-1谱段范围内特征性较强,作为建模的区域。详见表4。
主因子数选择:建立模型时所选的主因子数对预测结果会产生较大影响,所以在相同预处理方法及波段条件下,对其主因子数进行选择。因子数太小,样品模型代表性不强,模型覆盖面窄;因子数太大,则模型样品代表性杂乱。采用内部验证法,根据内部RMSECV、R2随主因子数的变化图,当因子数为10时,可得到最小验证集RMSECV和最大验证集 R2,故选择因子数为10。详见图3和图4。

表2 43批附子药材及炮制品的双酯型生物碱含量(HPLC法和近红外光谱法比较)
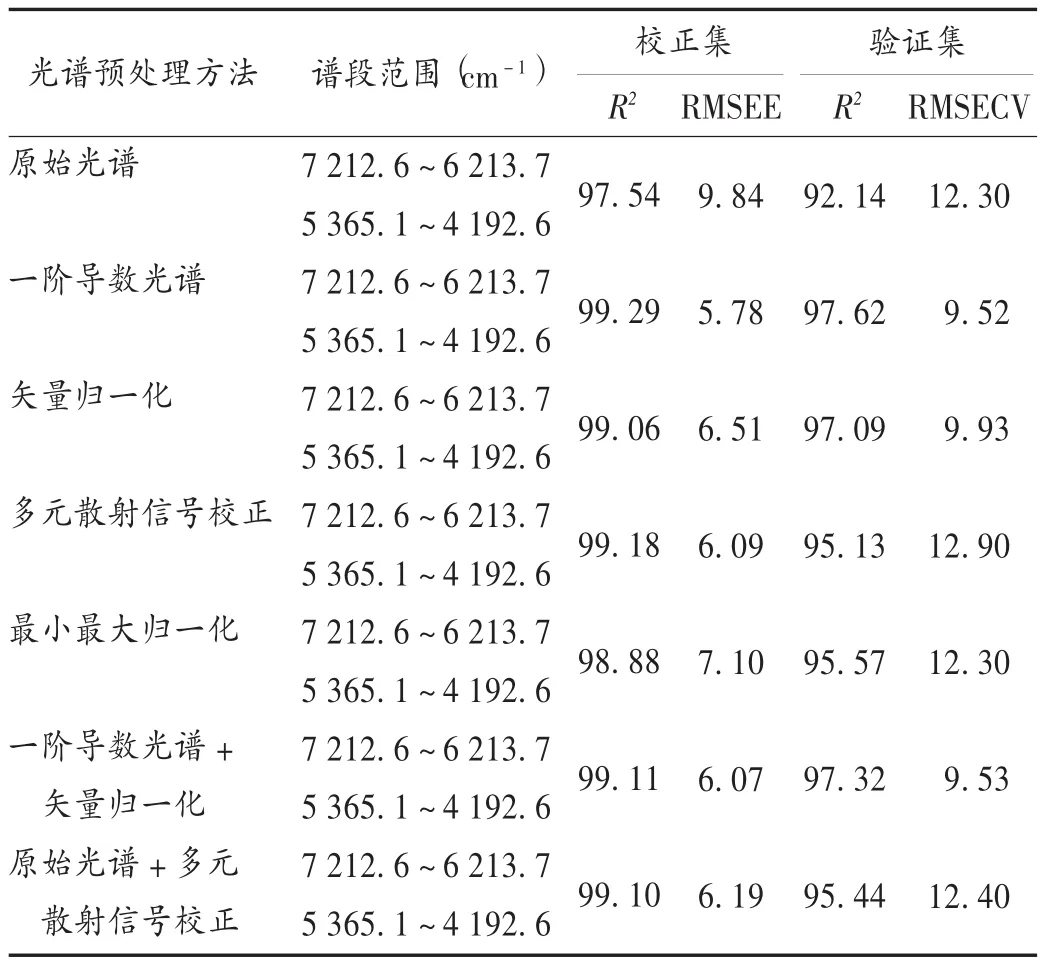
表3 双酯型生物碱光谱预处理方法选择

表4 双酯型生物碱谱段选择
2.4 定量模型的建立与验证
从50份样品中选择43个样品作为校正集、内部验证集,7个样品作为外部验证集,使验证集样品中的双酯型生物碱的含量在校正集含量之间,结果见表2。用样品的校正集建立校正模型,再作交叉验证。最后用验证集样品对模型进行外部验证,结果见表5。结果双酯型生物碱的 R2=98.24,RMSECV=8.18,说明该模型具有良好的线性和预测能力。

表5 7批附子药材及炮制品的双酯型生物碱含量(HPLC法与近红外光谱法比较)

图4 因子选择与验证集决定系数关系图
3 讨论
在研究中对样品粒度进行控制,可获得与HPLC法测量值接近的红外光谱图,使用附子及炮制品的原片与粉碎后样品的近红外光谱图有较大区别,附子及炮制品的粒度越大,光的折射就越多,光的漫反射就越小;反之,粒子越小,光就多为透射与折射,对样品特征信息的反映就越多。因附子及其炮制品在粉碎过程中易粘结,不易获得更细粉,选择过3号筛的粉能获得较好的NIR图谱。为使近红外收集到的信号平滑,消除偶然误差与噪声带来的影响,对收集到的样品扫描6次,取平均值参与建模。
附子炮制工艺不同,双酯型生物碱含量的差异较大。炮制不到位,临床用药安全难以保证;炮制过度,药效难以发挥。通过对四川与陕西两大产地的调研发现,附子的炮制加工方法不尽相同,在本地区时有毒性药材附子使用中毒的案例发生,因此建议监督部门对毒性药材附子在生产源头加强监管,使用部门严格按剂量使用,以防中毒事件的发生。
本试验中通过近红外光谱法与HPLC技术结合,采用偏最小二乘法建立了快速测定附子中双酯型生物碱含量的分析模型,结果表明,该方法准确、简便、快速、无污染,可实现大批量样品的快速分析,为毒性药材附子双酯型生物碱的在线检测与快速筛查提供了新思路和新方法。建立的检测方法既可用于中药生产与经营企业购进验收的快速初检与药材炮制的实时评价,又可作为各级监管部门对市场上附子及炮制品的快速筛查,有利于快速无损地发现问题产品,及早控制风险,降低检测成本,提高检测效率,对于打击制售假劣中药,净化药品市场,确保人民群众用药安全,具有非常重要的意义。
猜你喜欢
食品工业(2022年4期)2022-06-14
Digital Chinese Medicine(2020年3期)2020-12-14
中成药(2017年12期)2018-01-19
中成药(2017年12期)2018-01-19
中成药(2017年12期)2018-01-19
中成药(2017年7期)2017-11-22
中成药(2017年8期)2017-11-22
中成药(2017年4期)2017-05-17
浙江临床医学(2017年1期)2017-01-12
农家科技中旬版(2016年9期)2016-11-02
