
潜在类别因子模型在基于通路的稀有变异遗传关联研究中的应用*
2018-07-16 10:07广东药科大学公共卫生学院统计学教研室510310
中国卫生统计 2018年3期
广东药科大学公共卫生学院统计学教研室(510310)
冀晓慧 卜 涛 皮路程 赵 丽 刘 丽 李丽霞 郜艳晖△
【提 要】 目的 研究潜在类别因子模型(latent class factor model,LCFM)在基于通路的稀有变异遗传关联研究中的应用,并和潜在类别模型(latent class model,LCM)进行比较。方法 选取遗传分析工作组17(genetic analysis workshop 17,GAW17)中的VEGF通路变异数据及其200次模拟表型数据进行分析。将通路下基因中的稀有变异集合为一个新变量,和常见变异一起拟合LCM和LCFM,再调整年龄、性别和吸烟状态后分别拟合潜在类别及潜在类别因子对模拟表型Q1或Q4的线性回归模型(200次),并分别计算两种方法的统计效能和I类错误。结果 LCM将通路作为整体,统计效能为1.000,I类错误为0.030。LCFM将通路中的8个基因构造成三个因子,因子1和因子2含强效应基因,因子3含弱效应基因,其统计效能分别为0.980,1.000,0.595;I类错误分别为0.045,0.040,0.070。结论 LCFM比LCM更适宜分析通路数据。LCFM不仅能对通路中多基因信息降维,且能够根据后验概率从多个维度对人群分类,而且能估计因子载荷以反映基因间的关联强弱,为进一步的生物学机制研究提供线索。
全基因组关联研究(genome-wide association studies,GAWS)识别出的常见变异对疾病遗传风险的解释比例仍较低[1],近期很多研究证实低频或稀有变异和复杂性状存在关联[2],且具有很强效应[3]。二代测序技术使稀有变异的检测不再是难题,但也迫切需要相应统计方法的发展。稀有变异频率低,使用传统分析方法效能低下,因此统计学者提出负担检验[4](burden test)的策略,先对感兴趣区域(region of interest,ROI)如基因内多个稀有变异位点集合(collapsing),再比较病例组和对照组的遗传得分,或纳入回归模型同时分析稀有和常见变异或协变量。
虽然负担检验可提高基因内稀有变异关联分析的效能,但疾病的发生或改变通常是某些遗传通路中多基因共同作用的结果,且各基因效应可能不同[5]。因此如基于遗传通路,运用主成分或结构方程模型等潜变量(latent variable)方法[6],以更综合的角度解释遗传变异对疾病的影响。此外,由于遗传数据维度高且为分类变量,近年来以处理分类变量为优势的潜在类别模型[7](latent class model,LCM)也被用于遗传关联研究中,如分析通路数据时,多基因间的关联可由多个分类潜变量来解释,构建潜在类别因子模型[8](latent class factor model,LCFM)。实际应用中,LCFM可将多个基因位点归属为不同的潜在因子,进一步在回归框架下研究各潜在因子和表型的关联,为揭示多基因复杂的相互作用提供较为丰富的生物学线索。本文利用遗传分析工作组17(genetic analysis workshop 17,GAW17)中的通路数据,探讨结合负担检验的LCFM在稀有变异关联研究中的适用性,并和一般的LCM进行比较。
资料与方法
1.数据来源
本研究数据来自德克萨斯州医学研究中心GAW17[9],包括697例无血缘关系人群的3205个基因24478个SNPs(single nucleotide polymorphism)基因型及年龄、性别和吸烟状态等真实数据,其中SNPs数据中包含一条信号转导通路 (vascular endothelial growth factor,VEGF)信息;并假设该通路上8个基因67个SNPs中的38个SNPs与定量表型Q1关联,模拟了200个表型数据集。除Q1外,模拟表型中还包含不受数据库中任何SNPs影响的定量表型Q4。根据与Q1关联的假设,VEGF的8个基因当中,KDR、VEGFA和FLT1为强效应基因,ARNT、HIF1A和FLT1效应强度中等,HIF3A、ELAVL4效应强度较低。
2.潜在类别因子模型
LCFM在LCM的基础上结合了因子分析(factor analysis,FA)的思想,当群体的异质性由多个维度引起时,可用LCFM对异质性观测从多个维度进行分类,并研究潜变量间的关联[10]。设x1,x2,…,xL表示L个离散型潜变量,则显变量的联合概率为:
(1)
式(1)即LCFM,其中P(Yn)是多个潜变量分类下概率函数P(Yn/x1,x2,…,xL)的加权,而权重大小是其所属L个潜变量联合分布的概率。在LCFM 中,潜变量的联合分布概率之和等于1,显变量在给定潜变量条件下相互独立。
LCFM的参数估计同LCM,可采用最大似然法(maximum likelihood,ML)。模型评价可采用BIC(Bayesian information criterion)、AIC(Akaike information criterion)和对数似然函数LL等信息统计量。指标越小,模型拟合得越好。根据最优模型,LCFM构造多个潜变量来说明观测的后验类别属性,并根据每个潜变量各水平的后验概率大小,将观测分配到最大后验概率水平中。
3.模型比较策略
由于关联分析时有效应变异和无效应变异共同存在于分析集中,为更接近实际分析策略,本研究将通路VEGF中8个基因的67个SNPs全部纳入分析。先将同一基因上的稀有变异根据负担检验中的指示赋值法(indicator coding)[11](1表示有,0表示无)集合,再与常见变异一起分别拟合LCM和 LCFM,获得单个潜变量或多个潜在类别因子;接着调整年龄、性别和吸烟状态后分别拟合单个潜变量或多个潜在类别因子对Q1或Q4的线性回归模型(各200次),分别计算两方法的效能(根据Q1的200次回归模型结果)和I类错误(根据Q4的200次回归模型结果)。
4.软件
本研究中LCM和LCFM采用Latent GOLD 4.5,其他分析应用R软件。VEGF通路的基因功能注释应用KEGG(Kyoto Encyclopedia of Genes and Genomes;http://www.genome.jp/kegg/pathway.html)数据库及查阅文献获得。
结 果
1.LCM和LCFM的最优模型选择
表1显示对通路VEGF中遗传变异数据拟合LCM时,根据BIC(LL)选择2分类模型为最优模型;根据AIC(LL)选择4分类模型。由于BIC考虑了样本量大小,兼顾到模型简约性,最终选择2分类为最优模型。对于LCFM,根据 AIC(LL)选择三因子2类别模型为最优。最优LCM和LCFM的轮廓图见图1。
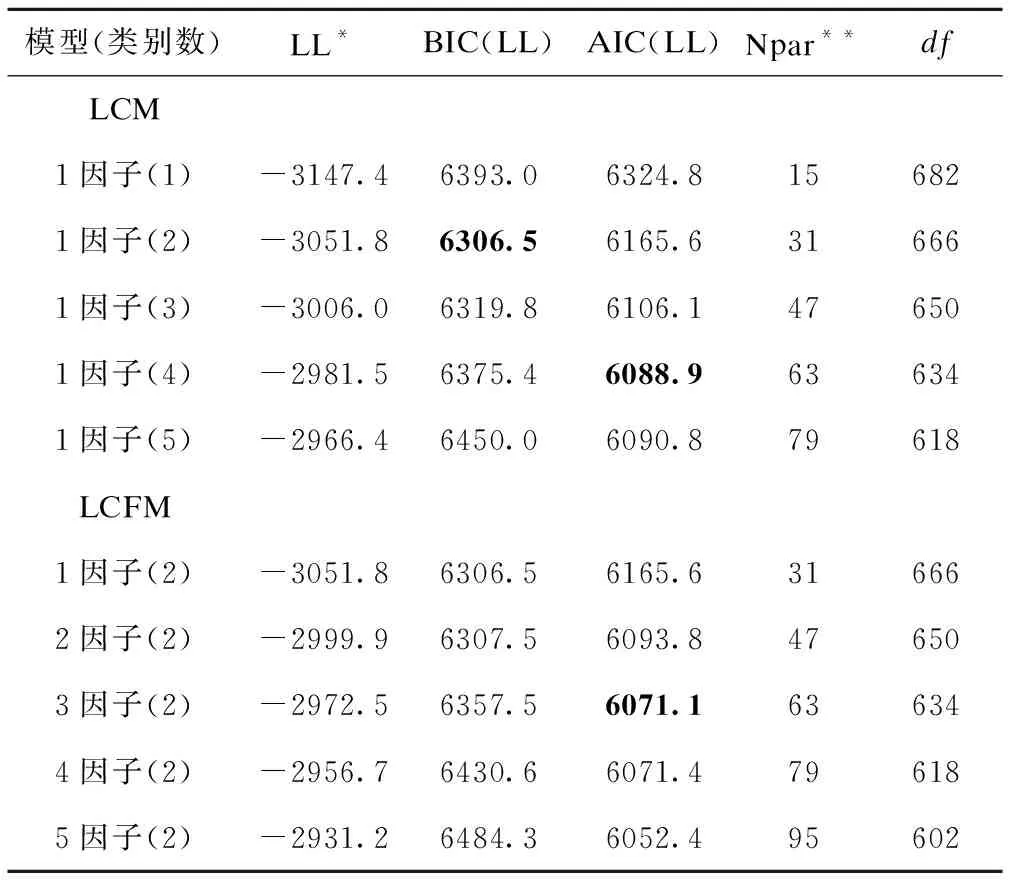
表1 Q1关联通路VEGF的LCM和LCFM最优模型选择
*LL:对数似然函数值; Npar:参数个数。
2.LCFM中VEGF通路各基因的因子载荷
表2显示LCFM中通路VEGF各基因因子载荷。因子1主要和KDR、VEGFA相关,因子2主要与FLT1、HIF1A、ARNT、FLT4相关;因子3主要与ELAVL4和HIF3A相关。
3.LCM和LCFM的效能和I类错误
基于LCM进行VEGF通路遗传关联分析时,通路作为整体,统计效能为1.000,I类错误为0.030。基于LCFM进行通路遗传关联分析时,通路中三个潜在类别因子的统计效能分别为0.980,1.000和0.595;I类错误分别为0.045,0.040,0.070。

注:RV表示稀有变异,CV表示常见变异
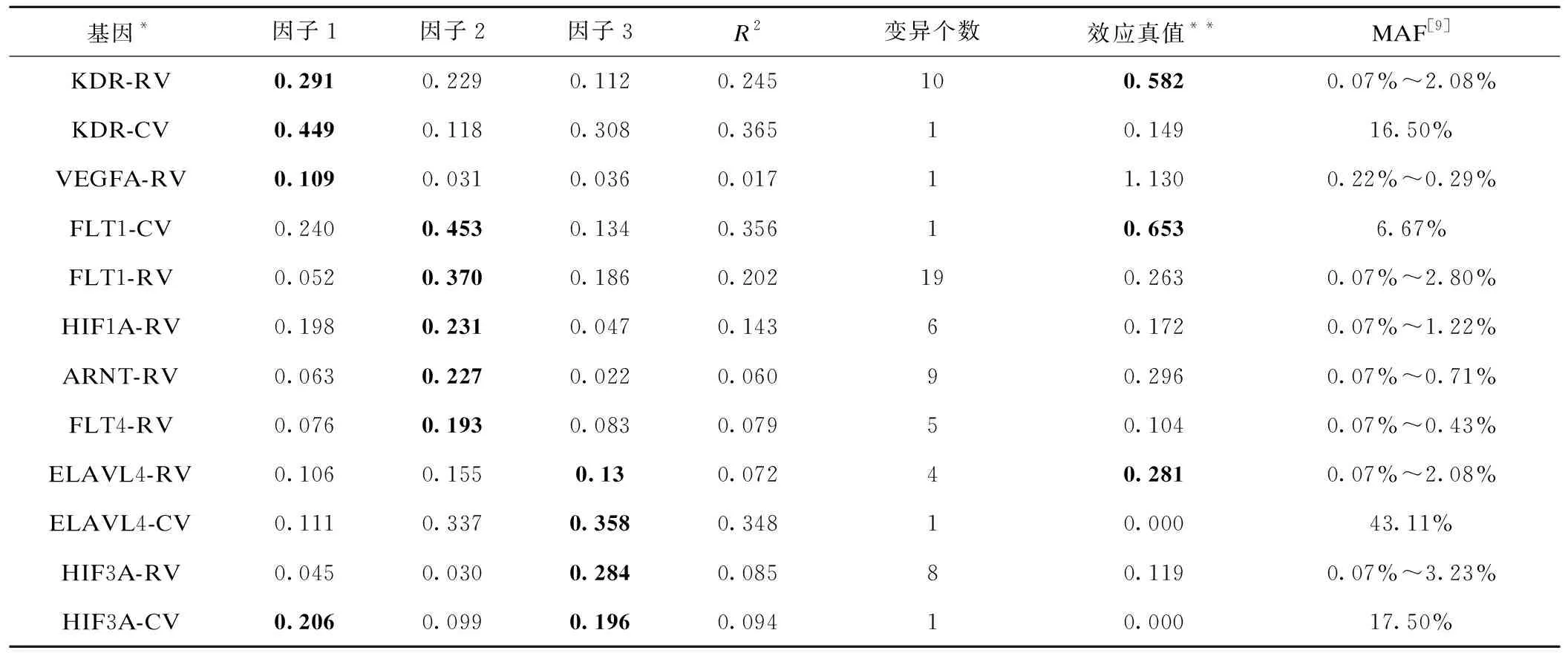
基因*因子1因子2因子3R2变异个数效应真值**MAF[9]KDR-RV0.2910.2290.1120.245100.5820.07%~2.08%KDR-CV0.4490.1180.3080.36510.14916.50%VEGFA-RV0.1090.0310.0360.01711.1300.22%~0.29%FLT1-CV0.2400.4530.1340.35610.6536.67%FLT1-RV0.0520.3700.1860.202190.2630.07%~2.80%HIF1A-RV0.1980.2310.0470.14360.1720.07%~1.22%ARNT-RV0.0630.2270.0220.06090.2960.07%~0.71%FLT4-RV0.0760.1930.0830.07950.1040.07%~0.43%ELAVL4-RV0.1060.1550.130.07240.2810.07%~2.08%ELAVL4-CV0.1110.3370.3580.34810.00043.11%HIF3A-RV0.0450.0300.2840.08580.1190.07%~3.23%HIF3A-CV0.2060.0990.1960.09410.00017.50%
*:RV表示稀有变异,CV表示常见变异;**:对稀有变异,效应真值为所有变异的效应均值。

表3 LCM与LCFM统计效能和I类错误
讨 论
虽然二代测序技术飞速发展,但单个稀有变异效应的检测功效极低,而基于由多个共同影响疾病特征的基因构成的通路分析策略,已被证实可提高遗传效应检测效能,并可从更全面的角度为潜在的疾病机制提供合理的生物学解释。本研究应用LCM和LCFM分析了GAW17中VEGF信号转导通路的遗传变异数据。其中LCM将通路中所有变异信息降维成1个分类潜变量;而LCFM构造了3个潜在类别因子,描述了多基因间的相关关系。基于潜在因子的遗传关联分析均有较高效能和较低I类错误。
已有研究表明,VEGF所介导的信号通路可调控血管内皮细胞的增殖、迁移和转化,促进血管新生,而缺氧是VEGF上基因转录的最主要诱导因子[12]。在缺氧诱导下,VEGFA等血管内皮生长因子大量产生,可与血管内皮细胞生成因子受体KDR结合,激活Src信号通路,引起内皮细胞发生和增殖,进而促进血管新生[13],而本研究LCFM中因子1主要与VEGFA和KDR有关。低氧时,缺氧诱导因子HIF1A(hypoxia inducible factor 1 A基)与1B基HIF1B,即ARNT结合形成异二聚体转录因子HIF1,并通过激活靶基因如酪氨酸激酶1,即FLT1 (fms-like tyrosine kinase 1)等调节靶基因的转录活性,引起血管形成。其中,FLT1促进血管细胞组成血管,而FLT4主要影响血管网络的构成[12],本研究LCFM中因子2主要与FLT1、HIF1A、ARNT和FLT4有关。HIF3A在调节缺氧的转录过程中扮演着重要监管角色,其大多数变体通过竞争性形成异二聚体而抑制HIF1A活性[14]。而ELAVL4与ELAVL1属同一家族,也可能有ELAVL1类似的功能作为基因转录后表达的重要调控因子[15],调控VEGFA、HIF1A mRNA 在细胞中的稳定性与翻译效率,从而调控靶基因在细胞中的表达水平[16],本研究LCFM中因子3主要与HIF3A和ELAV4有关。但是HIF3A和ELAV4在因子1和因子2中也有较高载荷,提示二者在调节转录和翻译过程共同促进或抑制基因表达。由此可见,LCFM各因子内的基因具有相关的生物学功能。
除将通路中多基因信息降维外,LCFM同时根据后验概率从多个维度将人群分类。应用于稀有变异数据时,LCFM可与负担检验的集合策略结合,提高了应用的广泛性。和LCM相比,LCFM更适宜分析通路数据。从生物学角度看,通路中多个基因或变异效应有强弱之分,且变异间可能存在结构或功能相似性以及复杂交互作用,但LCM可能将本属于不同功能的变异聚集为1个类别变量。而应用LCFM分析通路数据可能区别出不同功能的基因,且估计因子载荷以反映基因间的相关强弱,进一步通过遗传关联分析探索相关功能基因的整体效应,从而提高发现致病性基因或变异的效率,为进一步生物学机制研究提供线索。
实际应用中,除本文中采用的指示赋值法外,其他集合策略如比例赋值法(proportion coding)或加权合计检验(weighted_sum statistic,WSS)[11]等也可以和LCFM结合。由于稀有变异MAF越低其效应可能越强,因此根据MAF计算权重进行校正的集合方法可能提高研究效能。此外,本文利用了GAW17的模拟数据,仍需将LCFM应用到更多真实表型和测序数据中进一步验证其合理性和有效性。
猜你喜欢
新世纪智能(数学备考)(2021年9期)2021-11-24
少儿画王(3-6岁)(2020年4期)2020-09-13
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
当代陕西(2019年15期)2019-09-02
学苑创造·A版(2018年11期)2018-02-01
读者(2017年5期)2017-02-15
百科知识(2015年18期)2015-09-10
浙江大学学报(工学版)(2015年1期)2015-03-01
微型计算机(2009年4期)2009-12-23
