
噻吩在MoP表面的加氢脱硫反应实验设计
2018-07-03 07:11朱后禹匙玉华赵联明郭文跃
实验技术与管理 2018年6期
朱后禹, 徐 静, 匙玉华, 赵联明, 郭文跃
(中国石油大学(华东) 理学院, 山东 青岛 266580)
计算材料学(computational materials science)是材料科学与计算机交叉学科。该学科利用现代高速计算机技术,结合物理学、化学、生物学、材料学等相关学科的基础知识,可以对材料从微观到宏观多尺度的结构、性能等进行理论计算[1]。
计算机模拟技术可以模拟超高温、超高压等极端环境下的材料使用性能,模拟材料在使用条件下的性能演变规律和机理,进而实现材料使用性能的改善。因此,在现代材料学领域中,计算机实验已经成为与传统实验室的实验具有同等地位的研究手段[2]。 对于原子层次上的问题的求解,一般基于密度泛函理论(density functional theory,DFT)[3]的第一性原理(first principles)计算方法[4]。在现代石油工业中,加氢脱硫过程是一个重要环节,为此本文尝试对加氢脱硫反应从理论上进行实验设计。借助于Materials Studio程序包(Accelrys公司)中的DMol3模块进行计算。Materials Studio软件是一个跨越各种尺度的研究平台,不仅可以解析电子结构,还可以预测材料的宏观结构性能。实验采用了基于密度泛函理论的第一性原理计算方法,依托超算中心集群资源,通过模拟计算给出了噻吩在MoP表面的吸附几何结构、能量,以及脱硫反应的机理。基于学生所掌握的基本理论知识,选取难度适中的实验内容、适合计算材料学实验教学。
1 实验设计依据
由于石油燃料的输运过程的增加以及环境标准的提高,全球的石油领域工作人员一直在努力寻找一种可以更为清洁的开采使用能源的方法,脱硫反应便是其中的一个重点。石油炼化过程中,一个重要的脱硫方法便是加氢脱硫[5],它是石油化合物精炼的一个尤为重要的催化反应[6]。在脂肪族硫化物的加氢脱硫反应中,含钴和镍的二硫化钼(MoS2)催化剂的引入对其有很好的效果[7],但是对噻吩类物质效果不佳[8]。过渡金属磷化物在加氢脱硫反应中的催化作用在近几十年已经得到了广泛的研究[9]。研究发现,磷化钼(MoP)比传统的硫化钼在加氢脱硫反应中起到了更好的催化性能[10-11]。本实验应用Materials Studio软件,通过基于周期性密度泛函理论对噻吩在磷化钼不同表面上的吸附量化计算模拟,加深了学生对于吸附能、吸附位点、过渡态等概念的理解。
2 实验设计
2.1 模型建立
模型建立是模拟计算中最基础的一步,所构建的模型是否合理决定了实验是否可以正确进行。模型建立分为如下步骤:
(1) 首先在可视界面建模时从软件自带结构库中导出MoP晶胞;
(2) 利用软件建模工具进行切面、建立超晶胞、建立真空层等操作,综合考虑模拟真实性与计算精度、效率,超晶胞大小取3层厚度;
(3) 增加噻吩分子在MoP表面,此时需要考虑噻吩在MoP表面可能的各种位置,从而确定其表面最稳定的位置。
本次实验的所建的初始模型结构见图1。
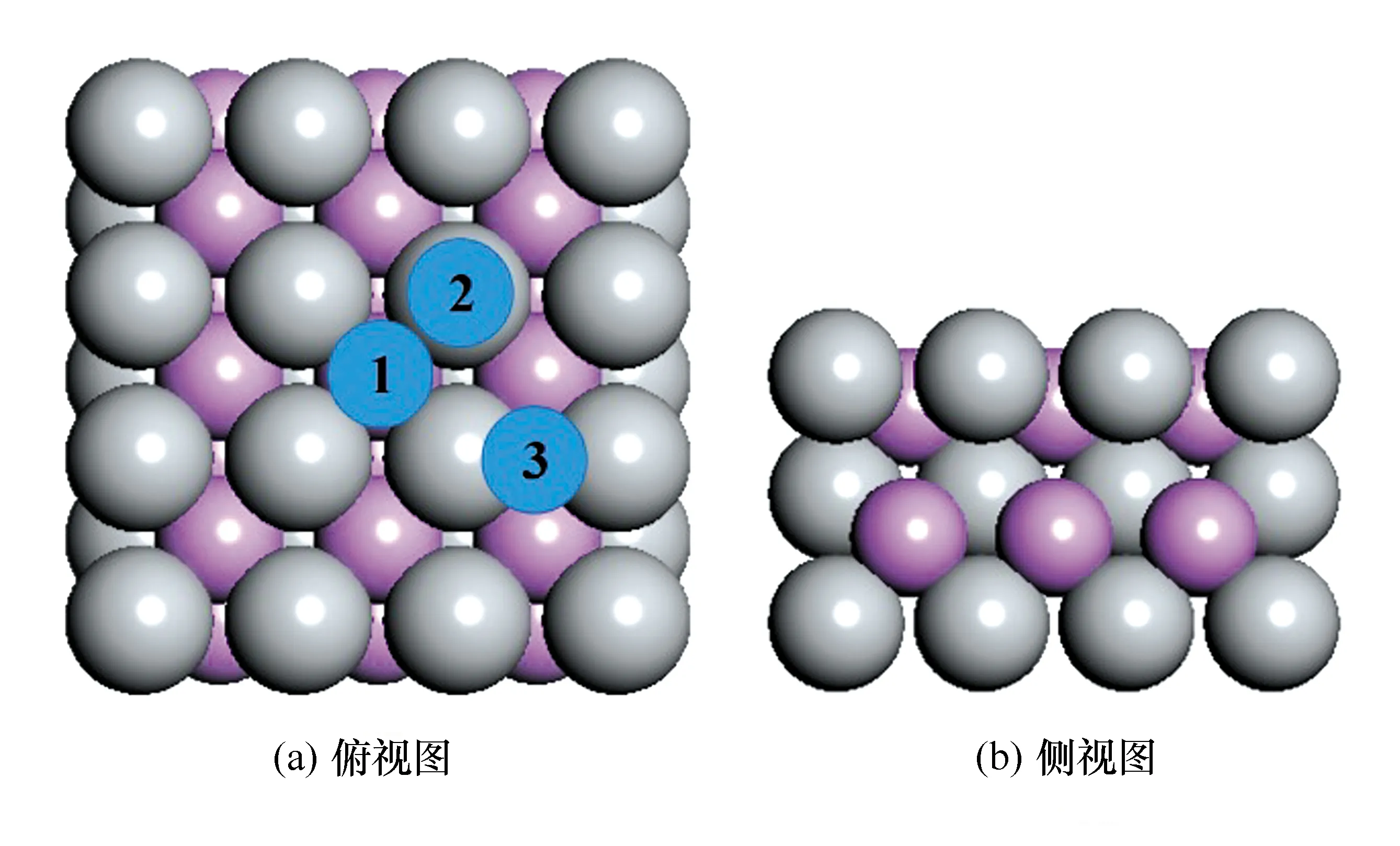
图1 MoP表面模型
2.2 模拟计算
本次实验的模拟计算包括模型优化和计算。优化计算得到的体相MoP的晶格参数:a=b=0.326 6 nm和c=0.319 8 nm,与实验结果(a=b=0.322 3 nm,c=0.319 1 nm)符合较好。后续计算也采用此参数以保证MoP能量保持在最低值。交换相关能以广义梯度近似(GGA)以及Perdew和Wang提出的(PW91)泛函计算[12-13]采用密度函数半核赝势(DSPP)方法描述金属原子的核内电子,用双极化基组加极化函数(DNP)处理价电子波函数,在DSPP优化结构基础上,采用全电子进行单点能量计算来校正磷化钼表面和吸附物质的电子性质[14-16]。所有的计算都用自旋极化进行。能量、位移和梯度的收敛标准分别为5.442×10-4eV,5×10-4nm和1.088 eV/nm。本次实验选取噻吩(T)在(001)表面上稳定吸附的T-bridge-fcc和(010)表面上稳定吸附的T-hollow-top构型分别开展计算。反应过渡态计算采用CI-NEB方法。
3 实验结果及讨论
3.1 噻吩在磷化钼(001)表面和(010)表面上的吸附构型
在MoP(001)上,噻吩有平躺吸附和垂直式两种类型的分子吸附,平躺吸附包括T-bridge-hollow(T-bridge-fcc和T-bridge-hcp)、T-hollow(T-fcc和T-hcp)和T-cross-bridge构型,图2示出了典型的吸附构型。为了研究在本结构中表面硫原子所起到的作用,计算了两种类型的表面硫原子,一种是S原子共享表面一个Mo原子,记为SS;另一种是C原子共享表面一个Mo原子,记为SC。在这种T-bridge-fcc构型中,噻吩分子中心在Mo2—Mo4桥位上方,噻吩S原子位于fcc位的上方,且分别与C2和C5共用Mo2和Mo4,另外2个C原子共用Mo3原子。在MoP表面,与金属表面比较,分子碳环平面倾斜了11°(图2(a))。在S-修饰的表面上,表面原子S和噻吩的共吸附结构见图2(b)和图2(c)。与在清洁表面上不同,形成一个新的S—Mo键,在硫修饰的表面上,噻吩中靠近表面S原子的钼原子的键长被拉伸了6.6%和3.1%,C2—S键缩短约为0.9%。同时观察到由表面硫原子引起的吸附物的移动。C—C和C—S键略微改变(C—C键0.2%,C—S键2.1%),噻吩在磷化钼(001)面上的吸附能,SS结构中减小8.5%和SC结构中减少21.2%。
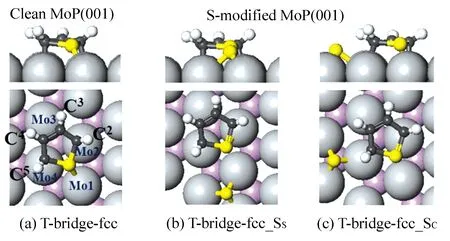
图2 在清洁和S-修饰的MoP(001)面上噻吩(T)的T-bridge-fcc吸附结构(C为黑色、S黄色、H白色、Mo浅灰色、P紫色)
在MoP(010)表面上,类似于MoP(001)的吸附情况见图3。噻吩环上的碳原子和噻吩下面的4个Mo原子的标记如图3(a)所示。为了研究表面S原子的影响,计算了两种类型的表面S原子,一种是S原子共享表面一个Mo原子,记为SS,另一种是C原子共享表面一个Mo原子,记为SC。在T-hollow-top构型(见图3(c))中,S原子位于Mo4原子的top位,C3位于Mo1和Mo2间的桥位之外,其余C原子位于邻近的Mo的top位。在清洁的磷化钼(010)上,噻吩的吸附形成了一个新的S—Mo键(0.248 2 nm)和4个新的C—Mo键(约0.232 0 nm),引起了C—S(约0.011 nm),C2—C3和C4—C5(约0.09 5 nm)键的较大程度的拉伸和C3—C4键(0.004 9 nm)轻微的伸展。此种吸附的吸附能为-2.55 eV。
在S-修饰的表面上,由于表面S原子和噻吩S和C原子共享Mo原子,结果T-hollow-top有两种稳定的共吸附构型,即T-hollow-top_SS和T-hollow-top_SC(见图3(b)和(c))。与清洁表面的情况相比,S修饰的表面因为形成了新的S—Mo键,涉及的噻吩的S/C—Mo键被拉伸0.001 4/0.002 4 nm,而噻吩的骨架键(C—C和C—S键)在0.000 1~0.002 3 nm的范围内变化。吸附能分别为-2.27和-2.28 eV,反映出表面硫原子减弱了噻吩在磷化钼(010)面上的吸附。
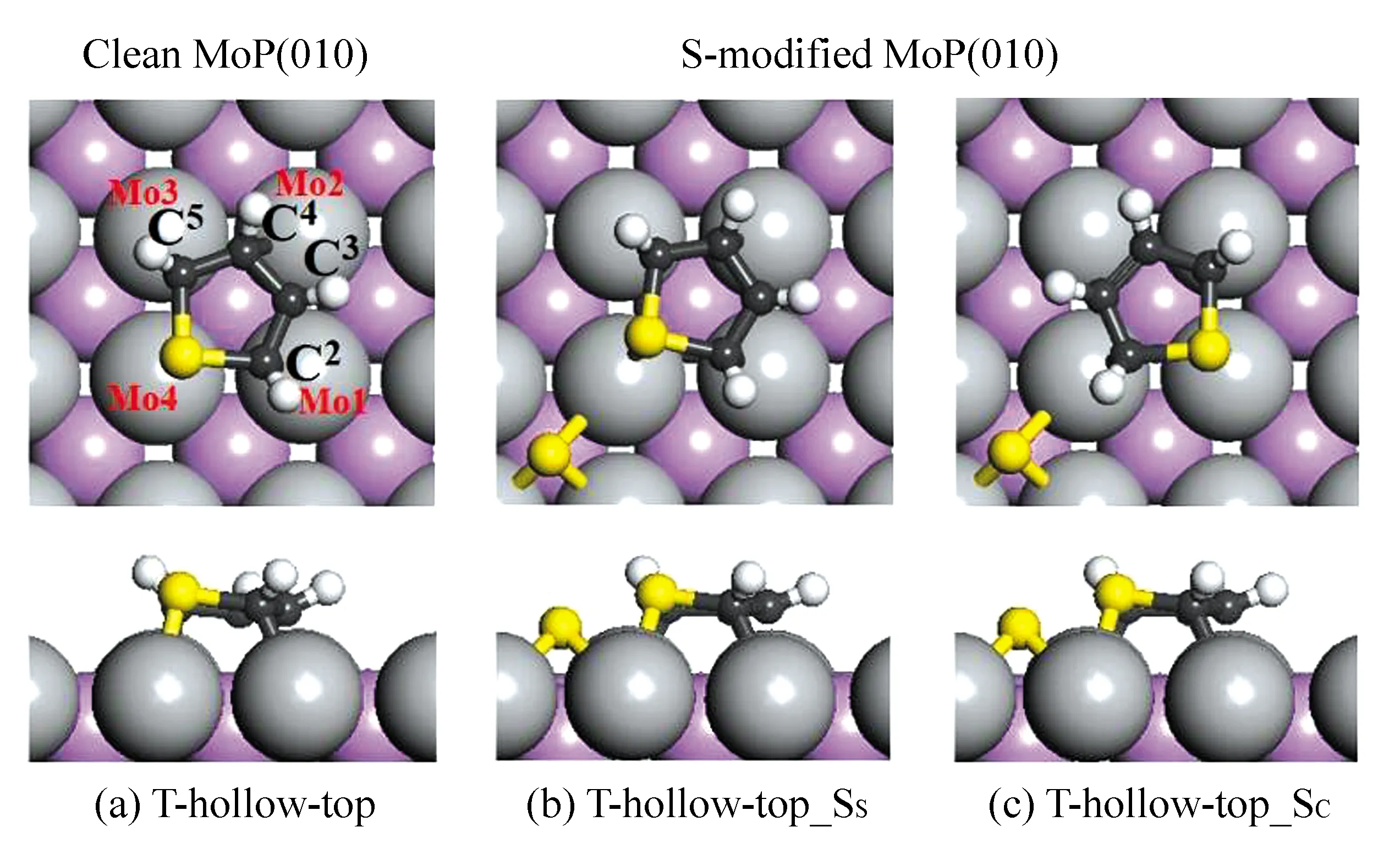
图3 噻吩(T)在清洁和S-修饰的MoP(010)表面上的稳定吸附构型(C原子黑色、S原子黄色和H原子白色)
3.2 噻吩在磷化钼(001)和(010)表面上的加氢脱硫反应机理
在MoP(001)表面,T-bridge-fcc脱硫的相关势能面(PES)以及所涉及的中间体的结构见图4。第一步,在TS1-A中,C2—S键断裂,反应能垒仅为0.18 eV,断裂距离为0.216 7 nm, C2—C3键经历了一个大的移动稍微偏向桥位,随后使C2原子移动到邻近的fcc位点,C3原子移到原来在IS中的C3和C4共享的表面Mo原子的顶部,形成了一个相当稳定的末态(FS),thiolate-A过程放热2.05 eV, C—S键断裂,组建一个有Mo原子的六元环,并且C—S键中插入了1个金属原子;第二步, TS2-A过程,C—S键的活化是由于C5—S键的伸缩振动,系统稳定1.31 eV,在FS((C4H4+S)-A)中,C5原子占据fcc位点,并且解离的S原子朝着相反的方向移向邻近的hcp位点,从而导致大的C5—S分离(0.482 6 nm)。在TS中,噻吩的S原子和C4—C5基位于IS和FS之间的中间位置,即噻吩的S原子位于桥位和C5原子位于表面Mo原子的顶位,且C5—S距离为0.262 0 nm。该过程活化能为0.44 eV。
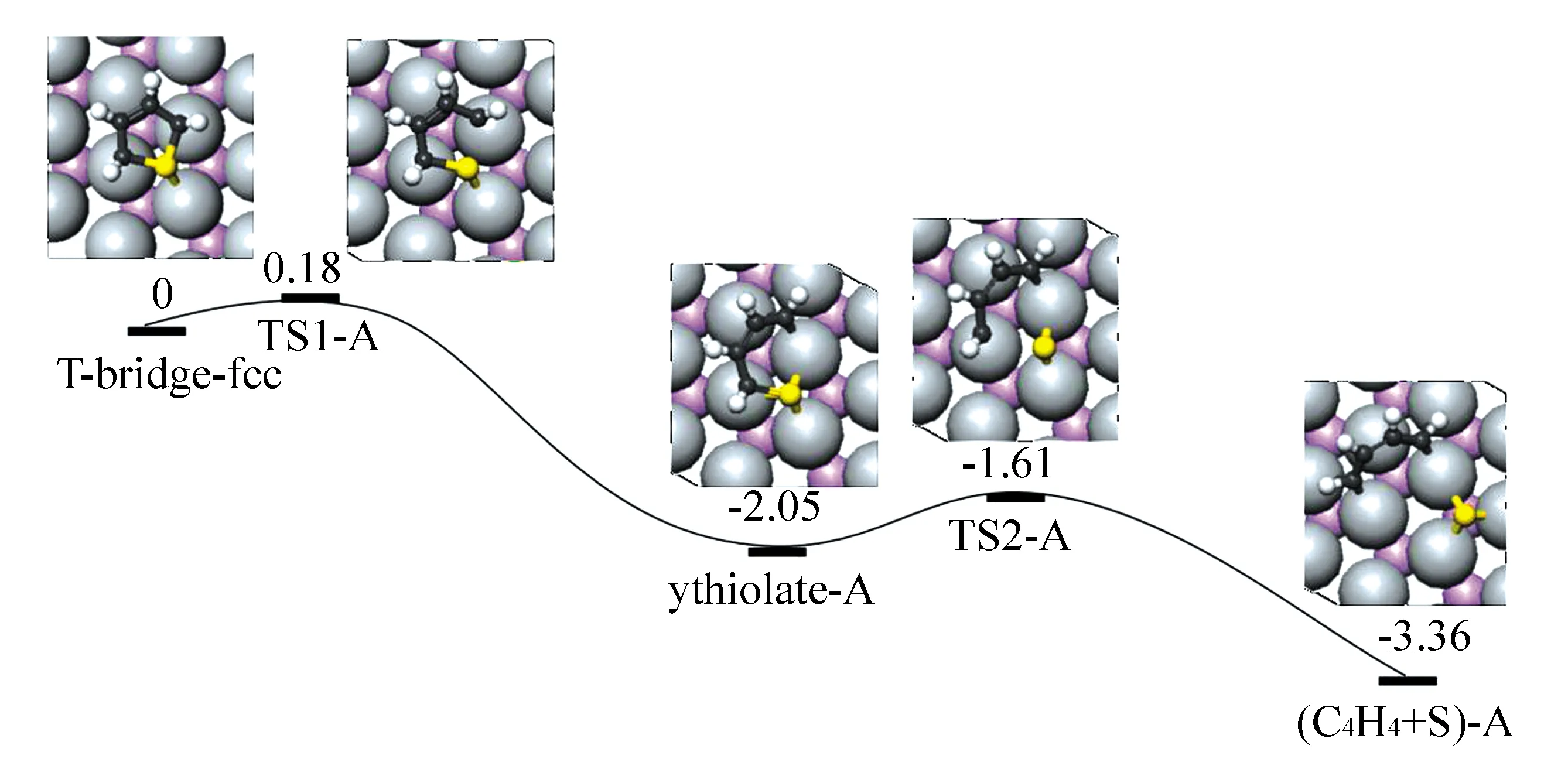
图4 MoP(001)表面上噻吩bridge-fcc构型的直接脱硫路径(T-bridge-fcc和吸附于无限远处2个H原子的总能量作为能量参考(eV))
图5给出了T-bridge-fcc构型的初始加氢路径以及相关结构。定义噻吩2号位(图5上面的路径)和噻吩3号位(图5下面的路径)的加氢路径分别为M和N。在过程M中,初始状态是邻近C2、C3的hcp位点的氢和bridge-fcc的位点的噻吩原子共同吸附。在过程TS-M中,氢原子向C2运动,H—Mo距离0.175 5 nm,C2—C3键偏向C2尾端的H原子, C2—S键断裂,断裂距离0.222 1 nm,C2—Mo键略拉长,C2与进攻的氢原子的间距是0.226 1 nm。过渡态后,C2—C3—C4角得到较大扩展,C2—C3键位于邻近fcc位点的顶点,且攻击的H与C2成键,最终C2—S分离(分离距离0.312 8 nm)达到稳定末态。此过程放热1.35 eV,能垒是0.63 eV。在IS-N构型里,攻击的H在C3原子的fcc位点上,与IS-M相比有点不稳定。在过渡态TS-N里,H到达与C3相邻的钼原子的顶点,H—Mo键距离0.182 1 nm。C3原子已经调整到便于攻击H原子的位置。这种结构预测1.48 eV的加氢能垒,在TS-N之后,3-MHT-bridge-fcc形成,该过程吸热0.82 eV。很明显,清洁表面上,噻吩2号位的加氢更具有优势。
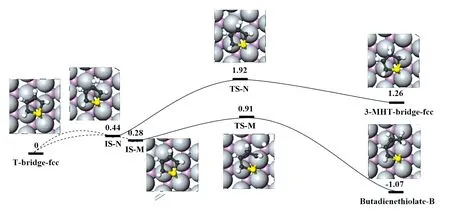
图5 MoP(001)表面上噻吩T-bridge-fcc构型的初始加氢路径(相应参数标注同于图4)
在清洁MoP(010)表面,T-hollow-top构型的脱硫反应发生了2个连续的C—S键断裂(见图6)。C2—S键断裂产生thiolate(C4H4S)-A,能垒为0.74 eV,反应能-0.42 eV。TS1-A中,S位移导致C2—S键断裂,C5—S键略微收缩(0.006 6 nm)。然后,稳定的中间体thiolate-A成为第一个C—S键断裂的终态(FS),S和C2保持在近邻bridge位,C2—S距离又被拉长。下一个C—S键断裂通过TS2产生稳定的FS((C4H4+S)-A),该系统比thiolate-A系统能量低0.32 eV。在TS2-A中,C5—S键已断裂,断裂距离0.248 3 nm,预测活化能垒0.55 eV。FS中,C4H4已各自移动到四重hollow位点,末端两个碳原子位于top和bridge处,解离的S位于bridge位。
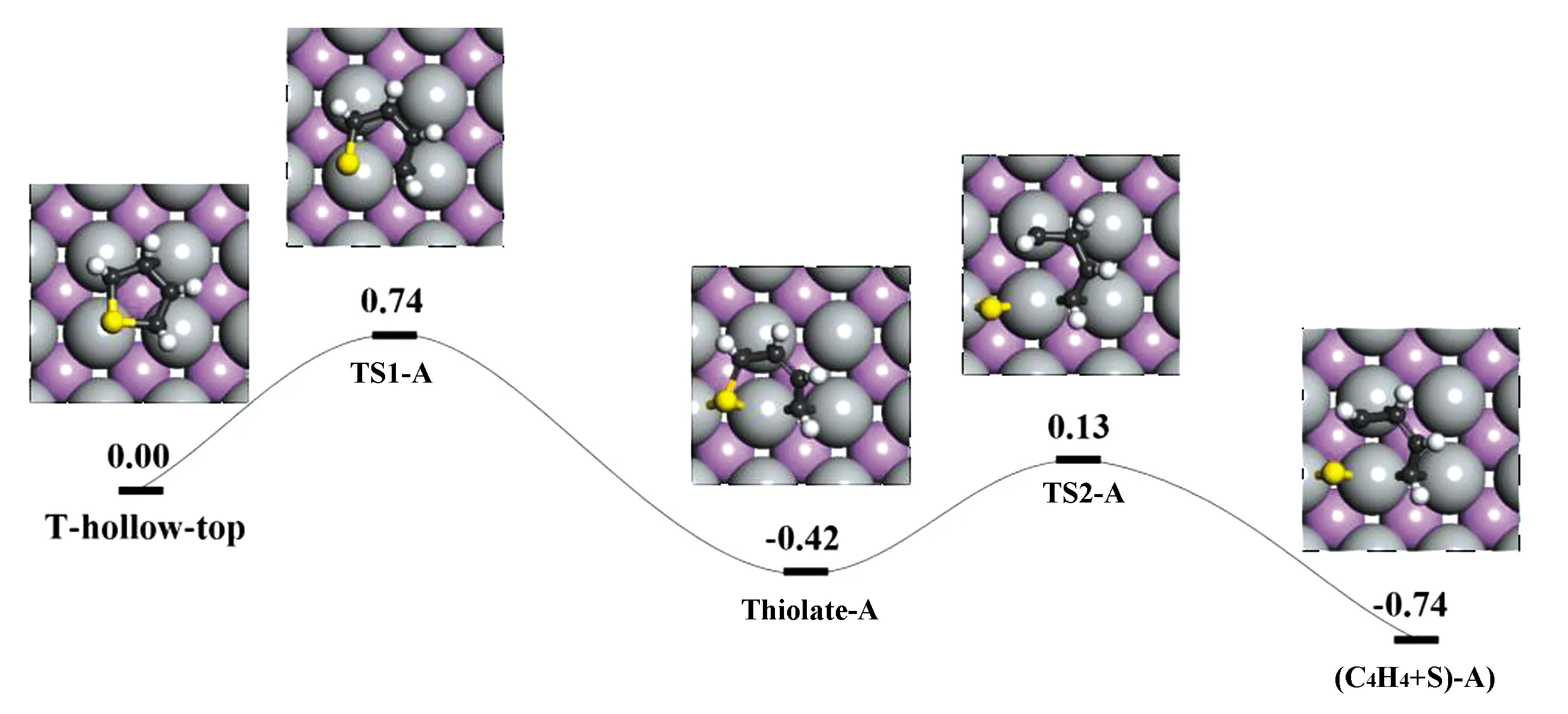
图6 MoP(010)表面上噻吩T-hollow-top构型的直接脱硫路径(T-hollow-top加上无限远处吸附的2个H原子总能量作为能量参考(eV))
对于T-hollow-top在清洁的表面上(见图7),在位置2和3处的加氢能垒为0.97 eV和0.98 eV,反应能各为0.46 eV和0.56 eV。

图7 MoP(010)表面上噻吩T-hollow-top构型的初始加氢路径
4 结语
在本实验的实施过程中,前期布置学生调研相关文献,调动学生主观能动性; 课上以学生为主体讨论实验步骤,分析实验结果;课后上交完整的实验报告。由于本实验依托于先进的超算中心计算平台,使得学生在掌握理论和实验知识的同时,也掌握了Windows和Linux双系统操作能力。该实验巩固了学生的专业基础知识,促进了理论与实践结合,提高了学生的科研素养。教学实践表明,学生更愿意接受这种参与性强、内容具有前沿性的研究型实验。
[1] 张跃,谷景华,尚家香,等. 计算材料学基础[M]. 北京: 北京航空航天大学出版社,2007.
[2] 坚增运,刘翠霞,吕志刚,计算材料学[M]. 北京: 化学工业出版社,2012.
[3] Hohenberg P,Kohon W. Inhomogeneous electron gas[J]. Phys Rev,1964(136):B864-B871.
[4] Segall M D,Lindan P J D,Probert M J,et al. First-principles simulation: ideas illustrations and theCASTEP code[J]. J Phys: Cond Matt,2002(14):2717-2743.
[5] Rodriguez J A, Kim J Y, Hanson J C, et al. Physical and Chemical Properties of MoP, Ni2P, and MoNiP Hydrodesulfurization Catalysts: Time‐Resolved X‐Ray Diffraction, Density Functional, and Hydrodesulfurization Activity Studies[J]. Cheminform, 2003, 34(39):1-8.
[6] Wang Huamin, Roel Prins. HDS of benzothiophene and dihydrobenzothiophene over sulfided Mo/γ-Al2O3[J] Appl Catal A: Gen, 2008, 350(2):191-196.
[7] Krebs E, Silvi B, Daudin A, et al. A DFT study of the origin of the HDS/HydO selectivity on Co(Ni)MoS active phases[J]. J Catal, 2008, 260(2):276-287.
[8] Yang R T, Hernández-Maldonado A J, Yang F H. Desulfurization of transportation fuels with zeolites under ambient conditions[J]. Science, 2003, 34(40):79-81.
[9] Oyama S T. Novel catalysts for advanced hydroprocessing: transition metal phosphides[J]. J Catal, 2003, 216(1):343-352.
[10] Phillips D C, Sawhill S J, Self R, et al. Synthesis, Characterization, and Hydrodesulfurization Properties of Silica-Supported Molybdenum Phosphide Catalysts[J]. J Catal, 2002, 207(2):266-273.
[11] Wu Z, Sun F, Wu W, et al. On the surface sites Of MOP/SiO2 catalyst under sulfiding conditions: IR spectroscopy and catalytic reactivity studies[J]. J Catal, 2004, 222(1):41-52.
[12] Perdew J P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas[M]// Density-functional approximation for the correlation energy of the inhomogeneous electron gas. New York: NRC Research Press,1986:8822-8824.
[13] Perdew J P, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Phys Rev B: Condens.Matter, 1992, 45(23):13244.
[14] Feng Z, Liang C, Wu W, et al. Carbon Monoxide Adsorption on Molybdenum Phosphides: Fourier Transform Infrared Spectroscopic and Density Functional Theory Studies[J]. J Phys Chem B, 2003, 107(49):13698-13702.
[15] Jun Ren, Chunfang Huo, Xiaodong Wen, et al. Thiophene Adsorption and Activation on MoP(001), γ-Mo2N(100), and Ni2P(001):Density Functional Theory Studies[J]. J Phys Chem B, 2006,110(45):22563.
[16] Li G, Zhu H, Zhao L, et al. Theoretical Survey of Thiophene Hydrodesulfurization Mechanism on Clean and Single-Sulfur-Atom-Modified MoP(001)[J]. J Phys Chem.C, 2016, 120(40):23009-23023.
猜你喜欢
电镀与精饰(2022年10期)2022-10-14
电镀与精饰(2022年3期)2022-03-14
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
表面工程与再制造(2019年6期)2019-08-24
北京航空航天大学学报(2017年10期)2017-04-20
当代化工研究(2016年1期)2016-03-16
合成化学(2015年10期)2016-01-17
新疆钢铁(2015年3期)2015-11-08
应用化工(2014年9期)2014-08-10
航天返回与遥感(2014年4期)2014-07-31
