
线粒体关联性内质网膜与细胞Ca2+依赖性死亡的研究进展
2018-06-21 01:13姚欢林育纯林忠宁
癌变·畸变·突变 2018年3期
姚欢,林育纯,林忠宁
(分子疫苗学和分子诊断学国家重点实验室,厦门大学公共卫生学院,福建厦门361102)
作为真核细胞极为重要的细胞器,线粒体和内质网(endoplasmic reticulum,ER)在细胞生命活动中紧密联系,已发现在线粒体外膜与内质网膜之间存在特殊的由特定蛋白质经由物理连接形成的亚细胞器结构,即线粒体关联性内质网膜(mitochondria-associated endoplasmic reticulum membranes,MAM)[1]。研究表明,MAM的功能与钙(Ca2+)信号调控、线粒体质量控制(mitochondrial quality control,MQC)、ER应激和脂质代谢等都有着密切联系[1];MAM已经被鉴定为细胞命运的调节枢纽,其调控机制与MAM功能蛋白和Ca2+信 号密切相关[2]。
1 MAM的生物学特性
2006年György等[3]采用透射电子显微镜技术,确证了线粒体和ER之间MAM区域保持稳定的膜间距,通常是25nm左右,不发生膜融合。MAM依赖其结构中作为物理连接的特定蛋白质组分,不仅为线粒体和ER相互作用提供了复杂多样的结构组成的空间基础,而且为各种信号通路相关蛋白质分子的“募集”或“组合”提供一个功能平台。由此,MAM结构及功能的协调稳定是维持细胞正常生理功能的物质基础,经由ER和线粒体的质量控制及其动态调节、以及二者Ca2+流变化、ER应激、线粒体相关性细胞凋亡等方面的调控,介导外源性环境因素诱导的细胞毒性、致癌作用、神经退行性变、代谢性疾病等细胞的转归和命运结局[1]。见图1。
1.1 MAM的结构组成
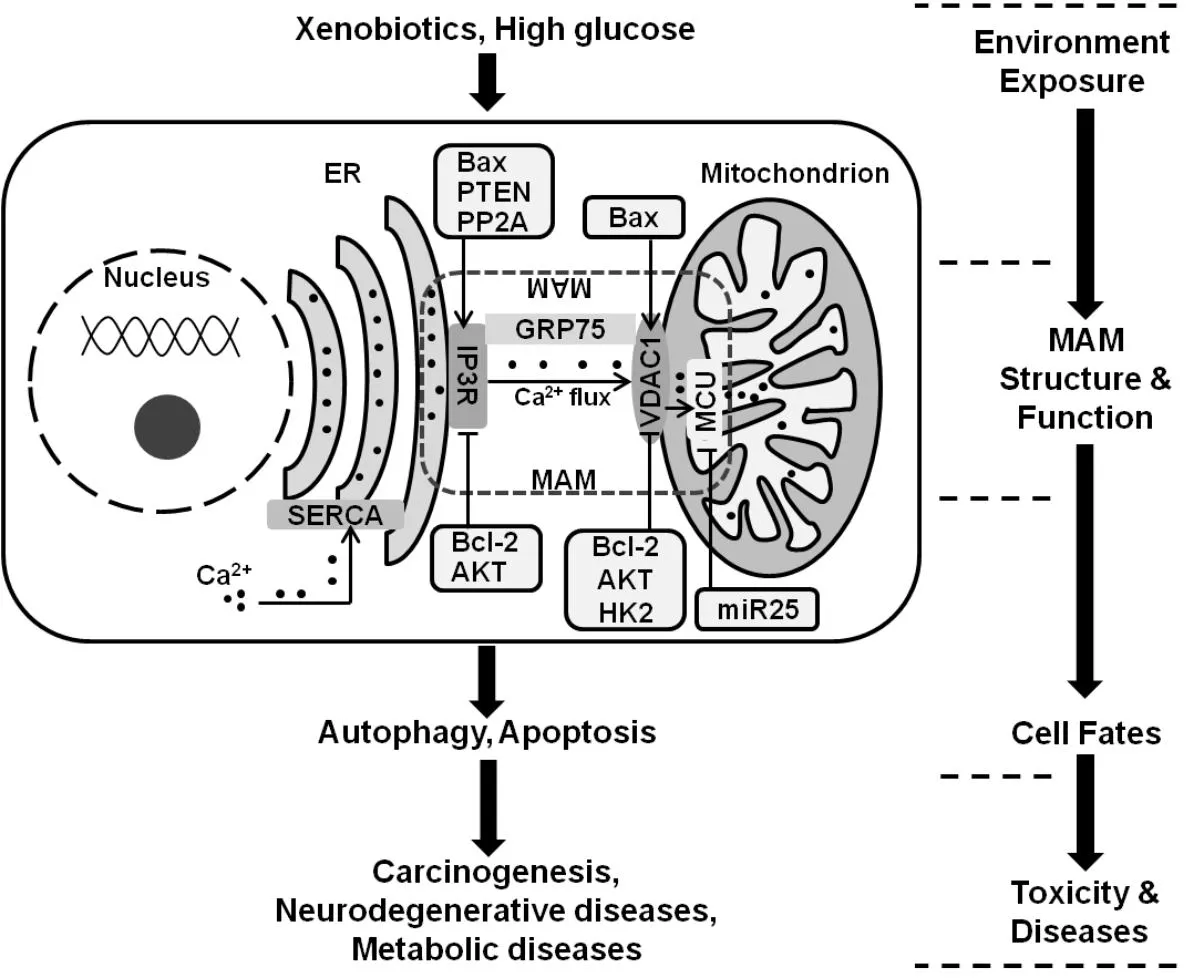
图1 MAM的结构组成及其功能
已有报告数十种蛋白质可分布或招募在MAM上,形成和维持其结构组成。MAM不同组分之间,可通过形成大的功能复合体、以及经由结构上或功能上复杂的相互影响,构成多样性功能的分子基础。已明确的,分子伴侣葡萄糖调节蛋白75(glucoseregulated protein75,GRP75)连接位于ER膜的钙释放通道1,4,5-三磷酸肌醇受体(inositol1,4,5-trisphosphatereceptor,IP3R)和线粒体外膜的电压依赖性阴离子通道1(voltage-dependent anion channel1,VDAC1)上,形成复合物在MAM的二个膜之间起到物理连接作用[4]。见图1。
随着MAM研究的深入,不断有新的蛋白组分被发现在MAM上,如内质网氧化还原酶-1α、蛋白磷酸酶2A(protein phosphatase2A,PP2A)、炎性小体NLRP3等被证实在MAM上发挥重要的物理连接作用。总之,MAM上这些各异的蛋白分子既是其物理结构组成的分子基础,又是其具体行使功能的承载者。已研究发现的MAM重要蛋白分子见表1;并且还有更多的MAM结构和功能相关蛋白需要进一步探究。
1.2 MAM的钙信号调控功能
MAM中多种蛋白与Ca2+信号调控相关,除IP3R、VDAC、GRP75等结构组分外,还包括钙联蛋白、MCU等特异性功能蛋白,见表1,共同形成有效的细胞器间Ca2+转 运机制[1]。ER外膜上IP3R的激活可使ER腔内Ca2+释放出来,在分子伴侣GRP75的帮助下,由线粒体外膜上Ca2+通道VDAC1摄入和经由线粒体内膜的MCU进入线粒体基质中。由此形成MAM中Ca2+信号流,经由ER和线粒体进行严格的调控,行使Ca2+信号转导功能,见图1。
1.2.1 MAM功能中的ER相关Ca2+信号调控ER是细胞内主要的Ca2+储 存库,是MAM功能性Ca2+流 的起始。胞浆Ca2+主要通过ER外膜上的SERCA进入ER,维持ER中正常Ca2+水 平[5]。SERCAs家族有 A TP2A1、 A TP2A2和ATP2A3三种不同的基因,各自转录和翻译成不同的变异体;其中SERCA2b具有最高的Ca2+亲和力,其功能对于ER正常的Ca2+摄取和细胞死亡的调节机制至关重要,是MAM行使Ca2+转 运功能的源泉[5]。

表1 MAM结构组成中的主要蛋白及其生物学作用[1]
ER的Ca2+可以通过尼丁受体(ryanodinereceptor,RyR)和IP3R两个通道释放到胞浆[6],二者是维持ER中Ca2+稳态不可缺少的另一类蛋白质;其中RyR主要在滑面内质网中表达(通常用做平滑肌和横纹肌细胞中的整个ER的标志物),其与细胞死亡之间的联系目前研究较少。IP3R则是在所有细胞类型中普遍表达的电导非选择性阳离子通道,是MAM的结构组成成分,其在细胞命运调控中的作用已有大量研究。IP3R通道存在3种不同的亚型(IP3R1、IP3R2和IP3R3),都可以被第二信使IP3、Ca2+、Ca2+结合蛋白和ATP激活,并且受硫醇修饰和磷酸化的几种蛋白(包括癌基因和抑癌基因编码蛋白)等的调控[7]。由此构成各种细胞MAM中Ca2+流转运的基础。
1.2.2 MAM功能中的线粒体相关Ca2+信号调控线粒体可以摄取从ER释放的Ca2+、 并储存少量的Ca2+以维持正常的功能。线粒体是MAM转运Ca2+流 的终点;Ca2+主要通过VDAC1进入线粒体外膜,到达线粒体膜间隙,然后通过位于内膜的MCU进入线粒体基质中(见图1),发挥生物功能[8]。
VDAC在人体细胞内有3个异构体(VDAC1/2/3), 其中VDAC1表达最多,在细胞凋亡中发挥重要作用,表现为VDAC1在MAM定位和选择性地与IP3R相互作用,从而增强凋亡Ca2+信 号转移到线粒体[2];并且MAM中的GRP75结构组分允许这个过程,而小干扰RNA(siRNA)沉默GRP75消除了IP3R和VDAC1之间的功能耦合,减少了激动剂(如组胺等)刺激后经由MAM 的线粒体Ca2+吸 收[4]。
为了使MAM的Ca2+流达到线粒体基质,位于膜间隙内部的Ca2+必须通过MCU复合体。MCU是一个成孔亚基组成的孔道复合物[9]。MCU的表达与MAM的Ca2+流最终进入线粒体基质发挥功能息息相关,其表达严格依赖于胞质Ca2+水平,并涉及核因子环磷腺苷反应元件结合蛋白,后者直接结合于M CU基因启动子而活化转录[10],由此调控MCU参与MAM的结构组分和Ca2+流的功能。
2 MAM与细胞Ca2+依赖性死亡
MAM相关Ca2+通 道在细胞Ca2+稳态调节功能中起着重要作用,它们活性的改变可能是细胞命运调节的关键原因。细胞Ca2+过载会引发活性氧(reactive oxygen species,ROS)产生的级联反应,导致线粒体膜电位下降和损伤。MAM蛋白通过绑定Ca2+以及调节Ca2+的 释放和摄取介导细胞内Ca2+的流动影响细胞死亡。MAM介导大量Ca2+转运可以促进细胞死亡,而且反应的强度和结果受存在于细胞器上的各种Bcl-2家族蛋白质调控(见图1)。抗凋亡成员Bcl-2和Bcl-XL抑制Ca2+转 运[11];促凋亡成员Bax/Bak、Puma和Bik刺激Ca2+转 运[12]。定位于线粒体和ER的Bax和Bak过表达促进凋亡相关Ca2+进程;而Bax/Bak双敲除小鼠胚胎成纤维细胞(mouse embryo fibroblasts,MEFs),则对Ca2+依赖性死亡刺激具有高度抗性[13]。
2.1 MAM相关ER钙泵与细胞Ca2+依赖性死亡
MAM中ER钙泵和Ca2+通 道控制的Ca2+流信号是生理和病理状况下决定细胞命运的主要因素;有两种主要的ER驻留蛋白参与这些过程。第一种是SERCA,其过表达不仅增加ER的Ca2+水平,而且在暴露于促凋亡物质(如HBV)时也增加了肝癌Huh7细胞的凋亡敏感性[14]。两个SERCA1剪接变异体编码的截短蛋白在Ca2+结合结构域的特征改变与ER中Ca2+稳 态水平降低和Ca2+释放增加有关,最终会导致Huh7细胞和宫颈癌HeLa细胞凋亡;而且SERCA1截短异构体的过表达会诱发HeLa细胞中的ER应激,并通过增加MAM偶联结构和线粒体Ca2+积 累来放大凋亡反应[14]。
MAM中另一种维持ER钙稳态的蛋白IP3R通过其表达水平和磷酸化的变化,调节Ca2+从ER转移到细胞内其他地方(特别是线粒体内),最终会改变细胞对死亡的敏感性。大量研究发现,MAM中IP3R在调控细胞命运和细胞死亡机制中起关键作用(见图1)。IP3R敲除的鸡淋巴瘤DT40细胞自噬增加,而IP3R稳定表达可完全恢复这些IP3R敲除细胞的基本自噬水平[15]。IP3R水平的降低会减少Ca2+转运,降低了线粒体代谢和ATP产生,从而触发MEFs细胞自噬。肿瘤抑制因子早幼粒细胞白血病蛋白通过影响IP3R活性来调节自噬反应[16]。PP2A通过去磷酸化IP3R,促进ER的Ca2+外排和MEFs细胞凋亡的发生[17]。原癌基因和抑癌基因编码蛋白通过调节IP3R表达和活性来发挥其作用,其中原癌基因编码的丝氨酸/苏氨酸蛋白激酶(RAC-αserine/threonineprotein kinase,Akt)介导以磷酸酶依赖方式抑制ER的Ca2+外排来调节人胚肾细胞(HEK-293)凋亡,而肿瘤抑制因子(phosphatase and tensin homolog deleted on chromosome ten,PTEN)通过去磷酸化IP3R来抑制Akt的作用,使得ER的Ca2+释放增加,促进HEK-293细胞凋亡[18]。靶向MAM的Ca2+转运可能是重建肿瘤细胞凋亡敏感性的有效方法,籍此开发的小肽BIRD-2 (Bcl-2-IP3受体抑制剂-2)可通过与Bcl-2结构域BH4结合,防止Bcl-2与IP3R的相互作用[11],诱导肿瘤细胞Ca2+介导的凋亡,并在小细胞肺癌细胞系中触发细胞死亡[19]。Foskett等[20]采用IP3R抑制剂XeB对MAM中IP3R介导的Ca2+流的抑制,使得乳腺癌细胞大量自噬死亡。
2.2 MAM相关线粒体钙泵与细胞Ca2+依赖性死亡
VDAC通道是线粒体摄取从ER释放的Ca2+通过线粒体外膜进入膜间隙的重要MAM组成部分;3种不同的VDAC亚型在哺乳动物组织中表达和显示相似的通道性质,但对线粒体Ca2+过载的凋亡敏感性具有相反的影响。
有研究发现,在大鼠成纤维Rat1a细胞中,Akt增加了己糖激酶2(hexokinase2,HK2)的活性,HK2可以和VDAC1相互作用,抑制VDAC1通道,阻止MAM Ca2+转运,减少了线粒体氧化磷酸化和细胞色素C的释放,从而抑制凋亡;在抑制Akt后,HK2与VDAC1分离,导致VDAC1打开促进MAM的Ca2+流并增加线粒体膜电位[21]。骨髓细胞白血病因子1可以与VDAC1相互作用,增加线粒体Ca2+摄取,从而促进肺癌细胞迁移[22]。Bcl-2家族的抗凋亡成员Bcl-xL与VDAC1相互作用,通过限制Ca2+信号转移到线粒体来保护MEFs细胞免受凋亡[8]。
与VDAC1不同,缺乏VDAC2的MEFs细胞更易发生凋亡[23];VDAC2与Bcl-x(S)相互作用并导致Bak释放而具有抗细胞凋亡作用[24]。研究发现,VDAC2特异性与Bak相互作用,VDAC2 过表达选择性抑制Bak激活、并抑制线粒体凋亡;抑制VDAC2使Bak从线粒体转移到细胞内其他区域(如过氧化物酶体膜)[25]。
MCU作为MAM中Ca2+流的最后通道,其亚基和功能的调节,会影响线粒体摄取Ca2+,产生过多ROS,从而增加肿瘤细胞发生凋亡的可能性;MCU激活通路会被钙调蛋白依赖性蛋白激酶依赖的磷酸化抑制,酪氨酸蛋白激酶可以阻止线粒体Ca2+过载、ROS产生以及随后的大鼠肝细胞凋亡[26]。组胺刺激HeLa细胞时,MCU表达的细胞线粒体Ca2+摄 取增加,导致凋亡[27];相反地,miR-25减少MCU表达,或者减少与MCU同型的内源性复合物MCUb的表达,它们则减少线粒体Ca2+摄 取,从而减少Ca2+依赖性的HeLa细胞凋亡[28]。
MAM上还有许多蛋白涉及凋亡的调节。内质网p53通过控制MAM中Ca2+转运来调节MEFs细胞凋亡[29]。在生长因子刺激MEFs中,定位在MAM上的雷帕霉素位点复合物2可磷酸化激活Akt,后者又磷酸化IP3R调节MAM的Ca2+功 能[30]。原癌基因编码蛋白H-Ras则可通过减少MAM的Ca2+转运,抑制小鼠胚胎成纤维3T3NIH细胞的凋亡过程[31]。
3 MAM结构和Ca2+功能调节的生物学作用
3.1 MAM调节异常的相关性疾病
研究表明,在许多疾病的病理过程中都伴随着MAM结构和功能的改变,MAM的Ca2+功能紊乱可能是某些疾病发生的重要指标。
MAM是肿瘤细胞代谢和进程的重要调节枢纽。有研究表明,与正常肝细胞相比,人肝细胞癌(hepatocellular carcinoma,HCC)细胞中MAM蛋白线粒体融合蛋白Mfn2表达降低,使得细胞对凋亡敏感性降低。而高表达Mfn2可以使HCC细胞中MAM的Ca2+转 运增强,诱发凋亡[32]。在HeLa细胞中,敲低MAM蛋白中定位于ER的硫氧还蛋白相关性跨膜蛋白1,会减少MAM接触位点,降低MAM的Ca2+转运,抑制线粒体代谢,加快肿瘤的生长[33]。丙型肝炎病毒p7蛋白可以促进人肝癌Huh7.5细胞MAM结构增强,导致MAM 的 Ca2+转运水平升高,引发线粒体Ca2+过载,促进了线粒体ROS产生,导致线粒体功能障碍从而产生代谢适应性反应,最终降低线粒体氧化磷酸化水平,增强糖酵解和脂肪生成[34]。
帕金森病(Parkinson’s disease,PD)与MAM的Ca2+功能失调有关。已知 α-突触核蛋白(α-synuclein, α-Syn)在PD的病理过程中具有重要作用;Cali等[35]通过在HeLa细胞中高表达 α-Syn,发现从ER到线粒体的Ca2+转运增加,证实 α-Syn通过增加MAM结构蛋白含量来调节Ca2+稳 态;线粒体Ca2+平衡的破坏,可能是MAM结构功能失衡和神经元功能紊乱的潜在因素。阿尔茨海默病病理过程中,也发现MAM上毒性淀粉样蛋白水平增高,导致MAM结构增强和Ca2+转 运水平升高[36]。亨廷顿氏病(Huntington’s disease,HD)的特征是突变的亨廷顿蛋白,其导致二硫键异构酶(protein disulfide isomerase,PDI)在MAM上积累并触发神经系统损伤;使用小分子物质LOC14调节MAM中的PDI水平,可显著改善HD疾病模型小鼠的运动功能、减弱脑萎缩和延长存活[37]。
MAM在代谢性疾病中也发挥重要作用。在瘦素缺陷型ob/ob肥胖小鼠中研究表明,肥胖促进了肝脏MAM结构形成,促使MAM的Ca2+转 运功能增强,造成线粒体Ca2+过载及功能障碍[38]。肥胖症和2型糖尿病模型小鼠的骨骼肌中显示出明显的MAM完整性破坏,这是线粒体功能障碍和胰岛素抵抗之前的早期事件;肥胖症患者肌管中胰岛素抵抗与MAM完整性破坏相关;表明MAM完整性破坏是小鼠和人类肌肉胰岛素抵抗相关的亚细胞改变[39]。MAM作为肝脏中激素和营养信号传导的重要枢纽,在高糖饮食情况下,可以破坏胰岛素抵抗 o b/ob小鼠肝脏MAM结构,干扰MAM的Ca2+转运功能,最终破坏糖代谢稳态;提示MAM介导的ER-线粒体相互作用通路参与代谢性疾病,并指向MAM作为代谢紊乱的潜在治疗靶点[40]。
3.2 MAM调节外源化学物诱导的细胞毒性
MAM在外源化学物暴露诱发的细胞或机体毒性损伤中有着极其重要的意义。Kyeong等[41]研究发现,人支气管上皮16HBE14o细胞暴露于二氧化钛纳米颗粒时,可诱导细胞ER应激,破坏MAM结构和Ca2+平衡,从而增加自噬。肾上腺皮质癌的最有效抗癌药物米托坦作用于人肾上腺皮质H295R细胞,破坏了MAM结构,造成MAM磷脂酰丝氨酸脱羧酶功能障碍,Drp1、乙酰辅酶A结合结构域3蛋白和转运蛋白的表达水平明显降低,最终诱发细胞凋亡[42]。在肝癌HepG2细胞中,棕榈酸可以破坏MAM结构,抑制MAM的Ca2+转运,从而增强细胞中线粒体ROS产量,抑制胰岛素信号;高表达Mfn2部分恢复了MAM 结构并改善了棕榈酸引起的胰岛素信号抑制[43]。这些研究表明MAM组成结构和功能的高度可塑性,是外源化学物作用的重要靶点,可能作为预防和治疗外源化学物诱发的细胞和机体损伤的干预靶点。
4 结语与展望
ER和线粒体作为重要的亚细胞结构,与细胞的功能状态密切相关;环境因素诱发的细胞毒性是造成机体损伤和疾病的主要原因;已有研究证明外源化学物可以靶向作用于细胞器,如线粒体和ER造成应激反应和损伤,特别是二者特化接触位点MAM的重要作用也越来越受到关注。MAM通过ER-线粒体结构偶联加强了两者之间的联系,在生理和病理状态下通过调节固定或招募在MAM上的蛋白组分和结构的动态变化,调控MAM的Ca2+转运功能,影响细胞的转归和命运。
已报道MAM中存在大量结构组成蛋白;外源化学物暴露的细胞中,定位于MAM的相关功能蛋白,如新鉴定发现MAM中环氧合酶2(cyclooxygenase2,COX-2)的分布,及其动态变化和功能学作用的调节,将为阐明MAM相关毒性(如致癌作用)机制和筛选生物标志提供新的突破;同时,探讨经由靶向干预MAM关键组分蛋白表达和修饰介导的细胞(器)损伤和毒性效应的病理过程调控,有望为探索细胞器之间接触位点的靶向干预,开展外源物(如环境诱变剂)暴露诱导毒性作用的评价和有害生物效应的预防控制策略提供新的视点。
[1]RATURI A,SIMMEN T.Where the endoplasmic reticulum and the mitochondrion tie the knot:the mitochondria-associated membrane(MAM)[J].Biochim Biophys Acta,2013,1833(1):213-224.
[2]DANESE A,PATERGNANI S,BONORAM,et al.Calcium regulates cell death in cancer:roles of the mitochondria and mitochondria-associated membranes(MAMs)[J].Biochim Biophys Acta, 2017,1858(8):615-627.
[3]CSORD Á S G,HAJN Ó CZKY G.Structural and functional features anDSignificance of the physical linkage between ER and mitochondria[J].J Cell Biol,2006,174(7):915-921.
[4]SZABADKAI G, BIANCHI K, VARNAI P, et al.Chaperonemediated coupling of endoplasmic reticulum and mitochondrial Ca2+channels[J].JCell Biol,2006,175(6):901-911.
[5]BRAVO-SAGUA R,RODRIGUEZ A E,KUZMICICJ,et al.Cell death anDSurvival through the endoplasmic reticulum-mitochondrial axis[J].CurrMolMed,2013,13(2):317-329.
[6]MARKS A R.Intracellular calcium-release channels:regulators of cell life and death[J].AmJPhysiol,1997,272(2Pt2):597-605.
[7]IVANOVA H,VERVLIETT,MISSIAEN L,et al.Inositol1,4,5-trisphosphatereceptor-isoform diversity in cell death anDSurvival[J].Biochim Biophys Acta,2014,1843(10):2164-2183.
[8]MONACO G,DECROCK E,ARBEL N,et al.The BH4domain of anti-apoptotic Bcl-XL,but not that of therelated Bcl-2,limits the voltage-dependent anion channel1(VDAC1)-mediated transfer of proapoptotic Ca2+signals to mitochondria[J].J Biol Chem, 2015,290(14):9150-9161.
[9]OXENOID K, DONGY, CAO C, et al.Architecture of the mitochondrial calcium uniporter[J].Nature,2016,533(7602):269-273.
[10]SHANMUGHAPRIYA S,RAJAN S,HOFFMAN N E,et al.Ca2+signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+uniporter geneMCU[J].Sci Signal,2015,8(366):ra23.
[11]RONGY P,BULTYNCK G,AROMOLARAN A S,et al.The BH4 domain of Bcl-2inhibits ER calcium release and apoptosis by binding theregulatory and coupling domain of the IP3receptor[J].Proc Natl AcaDSci U S A,2009,106(34):14397-14402.
[12]MATHAIJP,GERMAINM,SHORE G C.BH3-only BIK regulates BAX,BAK-dependent release of Ca2+from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death[J].J Biol Chem,2005,280(25):23829-23836.
[13]OAKES S A,SCORRANO L,OPFERMANJT,et al.Proapoptotic BAX and BAK regulate the type1inositol trisphosphatereceptor and calcium leak from the endoplasmic reticulum[J].Proc Natl AcaDSci U S A,2005,102(1):105-110.
[14]CHAMIM,GOZUACIK D,LAGORCE D,et al.SERCA1truncated proteins unable to pump calcium reduce the endoplasmic reticulum calcium concentration and induce apoptosis[J].J Cell Biol, 2001,153(6):1301-1314.
[15]CARDENASC,MILLER R A,SMITH I,et al.Essential regulation of cell bioenergetics by constitutive InsP3receptor Ca2+transfer to mitochondria[J].Cell,2010,142(2):270-283.
[16]MISSIROLI S, BONORAM, PATERGNANI S, et al.PML at mitochondria-associated membranes is critical for therepression of autophagy and cancer development[J].Cell Rep,2016,16(9):2415-2427.
[17]GIORGI C,ITO K,LIN H K,et al.PML regulates apoptosis at endoplasmic reticulum by modulating calcium release[J].Science,2010,330(6008):1247-1251.
[18]BONONI A,BONORAM,MARCHI S,et al.Identification of PTEN at the ER andMAM and its regulation of Ca2+signaling and apoptosis in a protein phosphatase-dependent manner[J].Cell Death Differ,2013,20(12):1631-1643.
[19]GREENBERG EF,MCCOLL K S,ZHONG F,et al.Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol1,4,5-trisphosphatereceptor disruptor BIRD-2and the BH3-mimetic ABT-263[J].Cell Death Dis,2015,6:2034.
[20]CARDENASC,MULLERM,MCNEAL A, et al.Selective vulnerability of cancer cells by inhibition of Ca2+transfer from endoplasmic reticulum to mitochondria[J].Cell Rep,2016,15(1):219-220.
[21]GOTTLOB K,MAJEWSKI N,KENNEDY S,et al.Inhibition of early apoptotic events by Akt/PKB is dependent on the first committeDStep of glycolysis and mitochondrial hexokinase[J].Genes Dev, 2001,15(11):1406-1418.
[22]HUANG H,SHAH K,BRADBURY N A,et al.Mcl-1promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+uptake and reactive oxygen species generation[J].Cell Death Dis,2014,5:1482.
[23]CHENG EH,SHEIKO TV,FISHERJK,et al.VDAC2inhibits BAK activation and mitochondrial apoptosis[J].Science, 2003,301(5632):513-517.
[24]PLOTZM,GILLISSEN B,HOSSINI AM,et al.Disruption of the VDAC2-Bak interaction by Bcl-x(S)mediates efficient induction of apoptosis in melanoma cells[J].Cell Death Differ, 2012, 19(12):1928-1938.
[25]HOSOI K I,MIYATA N.The VDAC2-BAK axis regulates peroxisomal membrane permeability[J].J Cell Biol,2017,216(3):709-722.
[26]GIACOMELLOM,DRAGO I,PIZZO P,et al.Mitochondrial Ca2+as a key regulator of cell life and death[J].Cell Death Differ,2007,14(7):1267-1274.
[27]DE STEFANI D,RAFFAELLO A,TEARDO E,et al.A fortykilodalton protein of the inner membrane is the mitochondrial calcium uniporter[J].Nature,2011,476 (7360):336-340.
[28]RAFFAELLO A, DE STEFANI D, SABBADIN D, et al.The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit[J].EMBOJ,2013,32(17):2362-2376.
[29]GIORGI C,BONORAM,SORRENTINO G,et al.p53at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner[J].Proc Natl AcaDSci U S A,2015,112(6):1779-1784.
[30]BETZ C,STRACKA D,PRESCIANOTTO-BASCHONG C,et al.Feature Article:mTOR complex2-Akt signaling at mitochondriaassociated endoplasmic reticulum membranes(MAM)regulates mitochondrial physiology[J].Proc Natl AcaDSci U S A, 2013,110(31):12526-12534.
[31]RIMESSI A,MARCHI S,PATERGNANI S,et al.H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling[J].Oncogene,2014,33(18):2329-2340.
[32]WANG W,XIE Q,ZHOU X,et al.Mitofusin-2triggers mitochondria Ca2+influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells[J].Cancer Lett,2015,358(1):47-58.
[33]RATURI A,GUTIERREZ T,ORTIZ-SANDOVAL C,et al.TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+flux[J].Cell Biol,2016,214(4):433-444.
[34]SCRIMA R, PICCOLI C,MORADPOUR D, et al.Targeting endoplasmic reticulum and/or mitochondrial Ca2+fluxes as therapeutic strategy for HCV infection[J].Front Chem,2018,6:73.
[35]CALI T,OTTOLINI D,NEGRO A,et al.Alpha-synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions[J].J Biol Chem,2012,287(22):17914-17929.
[36]VOLGYI K,JUHASZ G, KOVACS Z, et al.Dysfunction of endoplasmic reticulum(ER)and mitochondria(MT)in Alzheimer's sisease:therole of the ER-MTcross-talk[J].Curr Alzheimer Res,2015,12(7):655-672.
[37]ZHOU X,LI G,KAPLAN A,et al.Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington's disease[J].HumMol Genet,2018, 27(9):1545-1555.
[38]ARRUDA A P,PERS BM,PARLAKGUL G.Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity[J].NatMed,2014,20(12):1427-1435.
[39]TUBBS E, CHANON S, ROBERTM, et al.Disruption of mitochondria-associated endoplasmic reticulum membranes(MAMs)integrity contributes to muscle insulin resistance in mice and humans[J].Diabetes,2018,67(4):636-650.
[40]RIEUSSETJ.Endoplasmic reticulum-mitochondria calcium signaling in hepatic metabolic diseases[J].Biochim Biophys Acta,2017,1864(6):865-876.
[41]YU K N, CHANG S H, PARK SJ, et al.Titanium dioxide nanoparticles induce endoplasmic reticulum stress-mediated autophagic cell death via mitochondria-associated endoplasmic reticulum membrane disruption in normal lung cells[J]. PLoSOne,2015,10(6):e0131208.
[42]HESCOTS,AMAZITL,LHOMMEM,et al.Identifying mitotaneinduced mitochondria-associated membranes dysfunctions:metabolomic and lipidomic approaches[J].Oncotarget, 2017, 8(66): 109924-109940.
[43]SHINJO S,JIANG S, NAMETAM, et al.Disruption of the mitochondria-associated ER membrane(MAM)plays a central role in palmitic acid-induced insulin resistance[J].Exp Cell Res, 2017,359(1):86-93.
猜你喜欢
医学研究生学报(2022年5期)2022-12-07
临床肺科杂志(2022年3期)2022-11-26
中华实用诊断与治疗杂志(2022年1期)2022-08-31
保健医苑(2022年4期)2022-05-05
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
中国卒中杂志(2021年7期)2021-11-29
汽车维修与保养(2021年8期)2021-02-16
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
