
各Li吸附组分下硅烯氢存储性能的第一性原理研究∗
2018-06-14 06:31盛喆戴显英苗东铭吴淑静赵天龙郝跃
物理学报 2018年10期
盛喆 戴显英 苗东铭 吴淑静 赵天龙 郝跃
(西安电子科技大学微电子学院,宽带隙半导体技术国家重点学科实验室,西安 710071)
1 引 言
作为一种重要的类石墨烯二维材料,硅烯凭借与硅基半导体技术兼容的独特优势,在电子器件[1−3]、传感器[4]、催化剂[5]等领域受到密切关注.近年来,随着各国对氢能源的不断使用和重视,科研工作者开始利用硅烯较低的分子量和高比表面积的独特优势[6,7],研究硅烯在常温常压环境下作为氢存储材料的潜在可能性[8−10],从而避免使用传统高成本、高风险和较低存储密度的液化和加压的氢存储方式[9,11,12].研究发现,纯硅烯储氢能力很弱[13],吸附一个氢气分子的平均吸附能只有0.11 eV/H2,远小于美国能源部(U.S.Department of Energy,DOE)提出的5.50 wt%质量储氢密度和0.20—0.60 eV/H2平均吸附能的氢存储标准[6,9,13,14].但是由于硅烯具有sp2/sp3混合杂化的特性[15,16],在合适的金属修饰后,硅烯能够通过π键和金属原子紧密结合,并以金属原子为吸附中心吸附较多的氢气分子.这些金属原子也不会因为聚合而形成团簇[9,17],能够稳定分布在硅烯表面为氢气分子提供持久而有效的吸附环境,这一点对于金属修饰硅烯在氢存储领域中的循环使用尤为重要[11,18−20].
进一步研究发现,以Li,Na为代表的碱金属由于具备较低的内聚能[9,10,17]、较小的分子量[6,21]以及能够提供较为适宜的氢气吸附强度(介于强化学吸附和弱物理吸附之间)[22],比较适合用于氢存储领域中硅烯的修饰.Wang等[9]基于第一性原理计算,发现Li修饰硅烯(Li组分为0.20)中Li的结合能(2.69 eV/Li)远远大于其内聚能(1.71 eV),因此Li原子能够在硅烯表面稳定分布;进一步计算得到Li修饰硅烯的最大质量储氢密度为4.82 wt%,相应的平均吸附能为0.21 eV/H2,略低于DOE的氢存储标准,但也强于纯硅烯.Hussain等[22]利用第一性原理方法对Na修饰硅烯(Na组分为0.20)的储氢能力进行了研究,发现Na原子在硅烯表面也具有较大的结合能,并且每个Na原子能够吸附5个氢气分子,最终能够得到6.90 wt%的质量储氢密度和0.48 eV/H2的平均吸附能.显然,碱金属修饰硅烯能够有效提高硅烯的氢存储能力.但是,绝大部分科研工作者只是对单一组分下碱金属修饰硅烯的氢存储性能进行了研究,未曾探究不同组分碱金属对硅烯氢存储性能的影响,而后者可能对充分挖掘碱金属修饰硅烯的氢存储能力具有重要意义,并可为今后硅烯在氢存储领域的应用提供理论参考.
因此,基于第一性原理计算方法,本文从Li修饰硅烯入手,研究了不同Li吸附组分下硅烯体系(LixSi1−x)的结构和稳定性,讨论了各Li吸附组分下硅烯的储氢能力,并以质量储氢密度最高的Li饱和吸附硅烯为例,进一步讨论了Li修饰硅烯的储氢机理.结果表明,提高Li组分甚至使其达到饱和在理论上能使Li修饰硅烯具备更强的储氢能力,且电荷转移诱导的静电相互作用和轨道杂化作用是Li修饰硅烯储氢的关键.
2 计算方法
采用DMol3程序包[23]对硅烯进行第一性原理计算.其中,交换关联函数选择广义梯度近似(generalized gradient approximation,GGA)[24]下的Perdew-Burke-Ernzerhof泛函[25],基函数选择的是包含轨道极化函数的双数值轨道基组.此外,选用Grimme[26]提出的半经验DFT-D2方法修正范德瓦耳斯(van der Waals,vdW)相互作用,以获得较为可靠的库仑作用和氢气吸附能.计算过程中采用8×8×1的K点网格[27],5.1 Å的轨道截断半径.结构优化过程中,原子相互作用力阈值设为0.001 Ha/Å,原子移动的最大距离阈值设为3.0×10−3Å,体系中每个原子的能量收敛精度设为1.0×10−6Ha/atom.此外,为了研究各Li修饰硅烯体系中Li原子和硅烯表面的结合能力以及各Li修饰硅烯对氢气分子的吸附能力,定义Li原子的平均结合能(Eb)和氢气分子的平均吸附能(Eads)为:


其中E(silicene)和E(LixSi1−x)分别表示纯硅烯在m个Li原子吸附修饰前、后的总能量,x代表这m个Li原子所对应的组分大小;E(LixSi1−x−nH2)表示吸附n个氢气分子后Li原子修饰硅烯的总能量;E(Li)和E(H2)则分别表示相同晶胞尺寸下单个孤立Li原子以及单个孤立氢气分子的能量;m和n分别表示Li原子和氢气分子的数量.
3 结果与讨论
3.1 纯硅烯结构模型
硅烯是类石墨烯的二维薄膜材料,与石墨烯不同的是,其结构中具有2套不在同一平面的子晶格,因此组成翘曲的蜂窝状晶格结构,如图1所示.经结构优化,得到的原胞晶格常数为3.87 Å,最近邻Si原子之间键长为2.28 Å,两套晶格之间的垂直距离差(翘曲高度)h=0.46 Å,各参数结果和前人的报道相符合[6,7,13,22].为了最大化Li修饰硅烯的储氢量,选用单层2×2硅烯超胞(含8个Si原子)作为氢存储的研究对象,并且在考虑精度和效率的前提下,使用20 Å的真空层,以消除z轴方向上由周期性带来的影响.

图1 2×2单层硅烯超胞优化结构俯视和侧视图Fig.1.Top(upper panel)and side(bottom panel)views of the geometry structure of 2×2 supercell silicene.
3.2 不同Li组分下硅烯体系的结构优化及分析
3.2.1 优化结构分析
对于单个Li原子而言,在硅烯表面有四种典型的吸附位置,如图1中所示,分别为顶位(T)、谷位(V)、桥位(B)以及六元环中空位(H).先将1个Li原子分别放置在硅烯超胞表面的四种典型位置上进行结构优化,优化后的硅烯结构和对应的Li原子平均结合能分别如图2和图3所示.从图中可知,单个Li原子吸附在中空位时该硅烯体系(Li0.11Si0.89硅烯)具有最低的能量,相应的Li原子的平均结合能为2.11 eV/Li,说明中空位是单个Li原子在硅烯表面吸附的最稳定位置.值得注意的是,单个Li原子的平均结合能大于Li原子之间的内聚能(1.71 eV),可见该Li原子和硅烯之间存在较强的结合,能使Li0.11Si0.89硅烯即使在多次吸附和释放氢气后依然保持稳定.

图2 不同Li组分下硅烯体系的结构俯视和侧视图Fig.2.Top(upper panels)and side(bottom panels)views of geometry structures of LixSi1−x.
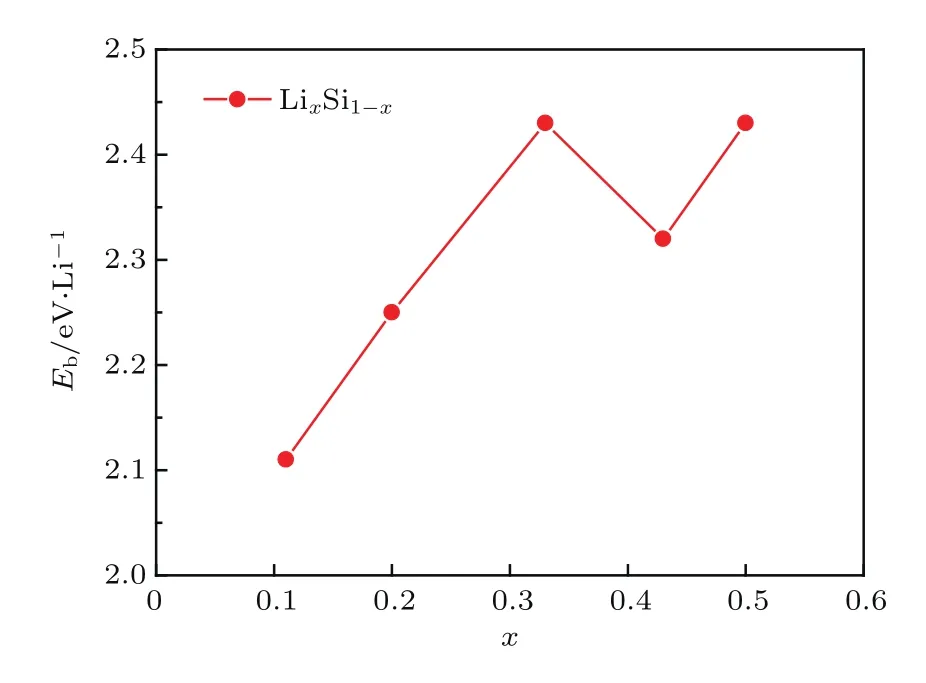
图3 不同Li组分硅烯体系中Li原子的平均结合能Fig.3.Binding energies of Li in LixSi1−x.
依次将2个、4个、6个和8个Li原子(分别对应Li0.20Si0.80,Li0.33Si0.67,Li0.43Si0.57和Li0.50Si0.50硅烯)放置在硅烯超胞表面各吸附位置并进行结构优化.从图2和图3中可以看到,Li0.20Si0.80硅烯中Li原子的最稳定位置依然是中空位,且超胞中的两个Li原子分别位于硅烯的上下表面,对应的平均结合能为2.25 eV/Li.在更高的Li组分下(Li0.33Si0.67硅烯),Li原子的最稳定位置转变到了谷位和桥位之间,并且具有2.43 eV/Li的平均结合能.随着Li组分的继续增加(Li0.43Si0.57和Li0.50Si0.50硅烯),Li原子的稳定位置逐步转向谷位,所对应的平均结合能分别为2.33 eV/Li和2.43 eV/Li.继续增大Li吸附组分,结构优化后发现Li原子的平均结合能大幅减小,并有低于其内聚能的趋势,因此本文认为当Li吸附组分为0.50时,Li原子在硅烯表面的吸附达到饱和,这也与前人的报道相符合[28,29].总体来看,随着Li组分从0.11增加到0.50,Li原子的平均结合能具有增大的趋势,且都能大于其内聚能,说明Li吸附组分在其达到饱和前的提高并不会给Li修饰硅烯的稳定性造成较大影响,后续对各结构进行氢存储研究是有意义的.
3.2.2 结合机理分析
为了理解Li原子和硅烯的结合机理,本文以Li吸附组分最高的Li0.50Si0.50硅烯结构为例,进行了Mulliken电荷布居和态密度分析.Mulliken电荷布居显示,由于Si原子相较于Li原子具有更大的电负性,每个Li原子向硅烯转移了约0.77个电子,从而使Li和Si原子分别变为带正电和负电的离子.带正电的Li离子和带负电的Si离子会形成一个较强的局域电场,这对于后续的氢气存储非常重要.同时,从图4的态密度图中可以看到,Si的s和p轨道与Li的s和p轨道产生了显著的杂化(−4.98—−0.51 eV以及0.60—6.23 eV),表明Li的s和p轨道参与了成键过程,同时也暗示了Li和Si之间存在着电荷转移.Li原子的p轨道也参与了成键,这是因为在Li原子和Si原子轨道杂化的过程中,Li原子首先向Si原子贡献了自己的s电子,导致部分Si原子的p轨道被占据;随后,周围Si原子产生的配位场使空的Li原子p轨道发生劈裂,并将一部分电子贡献回Li的p轨道,最终使得Li和Si之间存在较强的s-p和p-p轨道杂化[30].此外,由于Li原子的存在,Li0.50Si0.50硅烯结构产生了约0.7 eV的能隙,因此该结构具有半导体特性.

图4 Li0.50Si0.50硅烯结构态密度图Fig.4.Diagram of density of states of Li0.50Si0.50.
3.3 不同Li组分下硅烯体系的氢存储研究
3.3.1 最大储氢量判断和结构分析
基于上述优化后的硅烯结构,对各Li组分下硅烯超胞结构的氢存储性能进行了研究.我们将氢气分子以不同的位置和方向逐个添加在Li原子周围,借助结构优化来判断该硅烯体系的最大储氢量.通常情况下,Li修饰硅烯一旦达到了最大储氢量,后续添加的氢气分子在结构优化后会被排斥而远离Li原子,以保证硅烯体系的总能量最低[20].因此,定义氢气吸附距离为氢键中心点到Li原子之间的距离,并通过比较各氢气分子和第一个吸附在该Li原子上的氢气分子的吸附距离来判断硅烯体系是否已经达到最大储氢量.本文以Li组分最低的Li0.11Si0.89硅烯结构为例,介绍硅烯的最大储氢量判定过程(图5(a)).在这一过程中,我们还统计了该硅烯结构在吸附氢气过程中各氢气分子的吸附距离,如图5(b)所示.从图5(a)和图5(b)中可以看到,只添加了1个氢气分子时,该氢气分子在结构优化后倾斜地吸附在Li原子上,相应的吸附距离为1.96 Å;当添加了2个氢气分子时,结构优化后发现,这2个氢气分子在吸附后组成了一个俯视图上近似线性的结构,相应的吸附距离分别为2.01 Å和2.02 Å;当添加3个氢气分子时,各氢气分子的吸附距离在结构优化后增加到了2.05 Å,最终组成一个俯视图上近似对称的三角结构;继续添加氢气分子,结构优化后发现添加的氢气分子被排斥而远离Li原子,相应的吸附距离变为3.96 Å,远大于第一个氢气分子的吸附距离.可见,硅烯中的单个Li原子最多能吸附3个氢气分子,这与Wang等[9]的研究结果相同.因此,Li0.11Si0.89硅烯结构的最大质量储氢密度为2.54 wt%,相应的平均吸附能为0.58 eV/H2.
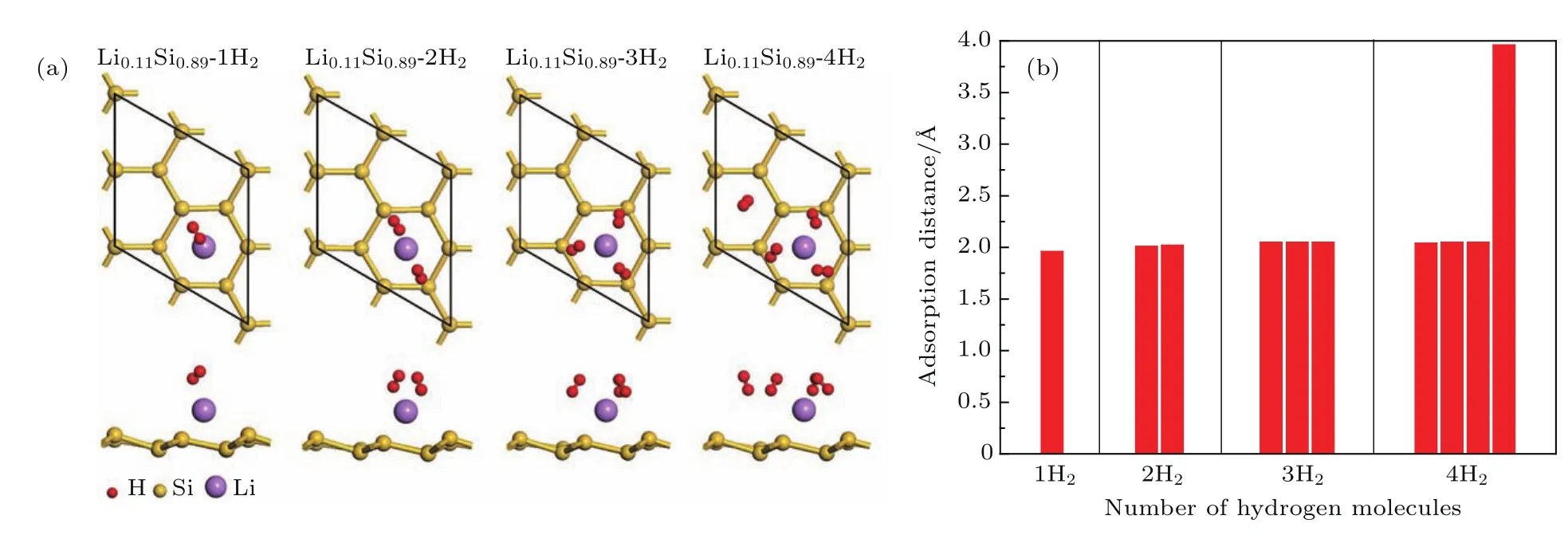
图5 Li0.11Si0.89硅烯最大储氢量判断的(a)结构图,(b)氢气吸附距离统计图Fig.5.Diagrams of(a)geometry structures with(b)corresponding adsorption distance statistics of Li0.11Si0.89 during the maximum hydrogen storage judgment.
根据上述最大储氢量判定方法,本文继续对其他硅烯结构的最大储氢量进行了研究.由于硅烯中的每个Li原子最多能吸附3个氢气分子,因此我们在考虑各硅烯结构的氢气吸附构型时,在每个Li原子附近摆放3个氢气分子,摆放的初始位置和方向与Li0.11Si0.89硅烯中的氢气分子类似,并且尽量使相邻Li原子吸附的氢气分子之间保持一定距离.经过多次氢气分子的位置、方向调整和结构优化,最终以未被排斥远离Li原子的氢气分子数量作为该硅烯体系的最大储氢数量.最终各硅烯体系的最大储氢后结构和氢存储性能可分别见图6和表1所列.从图6和表1中可以看到,随着Li组分的不断增加,储氢量也不断上升,表明提高Li吸附组分能够有效提升Li修饰硅烯的氢存储能力.Li0.20Si0.80,Li0.33Si0.67,Li0.43Si0.57和Li0.50Si0.50硅烯结构分别最多能够吸附6个、8个、14个和18个氢气分子,计算获得的最大质量储氢密度分别达到4.82 wt%,6.00 wt%,9.58 wt%和11.46 wt%,相应的平均吸附能分别为0.47 eV/H2,0.54 eV/H2,0.41 eV/H2和0.34 eV/H2.从表1中还可以看到,在计算中单独使用GGA会严重低估氢气分子的平均吸附能,而使用范德瓦耳斯修正能有效改善该问题,这与前人的报道相符合[8,14,22].最终可以发现,Li0.33Si0.67,Li0.43Si0.57和Li0.50Si0.50硅烯结构的最大质量储氢密度和平均吸附能都达到了DOE制定的储氢标准,能够作为氢存储材料使用,具有潜在的应用前景.
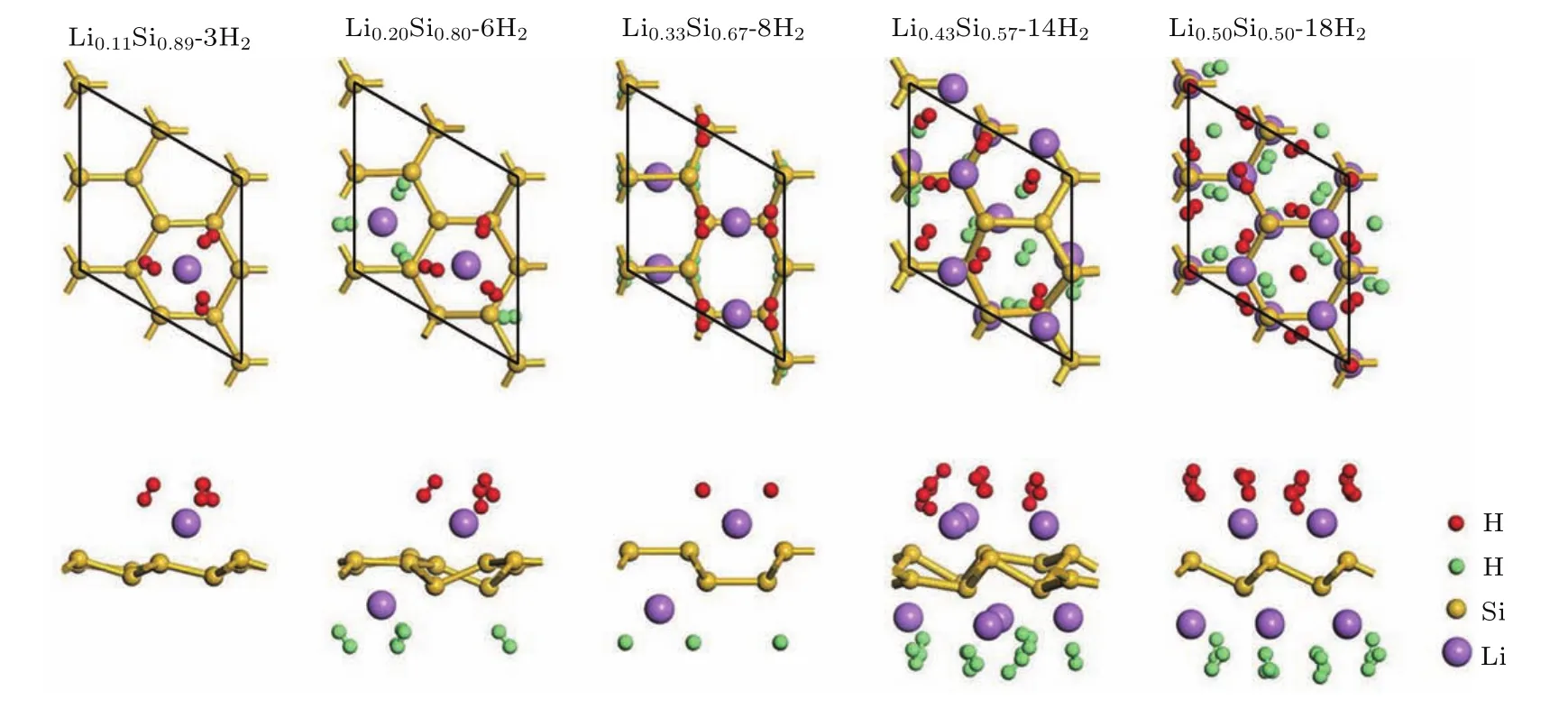
图6 不同Li组分下硅烯体系的最大储氢结构俯视和侧视图Fig.6.Top(upper panels)and side(bottom panels)views of geometry structures of LixSi1−xwith the maximum hydrogen storage.

表1 各硅烯体系的氢存储性能Table 1.The hydrogen storage properties of LixSi1−x.
3.3.2 储氢机理分析
为了探究Li修饰硅烯结构的氢吸附机理,本文以储氢量最大的Li0.50Si0.50-18H2硅烯结构为例,对Mulliken电荷布居、差分电荷密度和态密度进行了分析.从Mulliken电荷布居可知,随着Li0.50Si0.50硅烯不断吸附氢气分子,Li原子不断失去电子,每个Li原子失去电子的数量从0.77个电子单调增加到1.15个.这一失去电子的单调过程会使电荷转移形成的局域电场(Li正离子和Si负离子)不断加强,并通过极化作用吸附更多的氢气,这在差分电荷密度图中可以清楚地看到(图7(a)).黑色虚圆内的氢气分子靠近Li原子一侧的电荷密度增加(橙色),而远离一侧的电荷密度减少(蓝色),从而说明该氢气分子在局域强电场作用下被极化而形成了局部电偶极矩.因此,Li,Si离子同极化氢气分子之间的静电相互作用是Li修饰硅烯吸附氢气的原因之一.
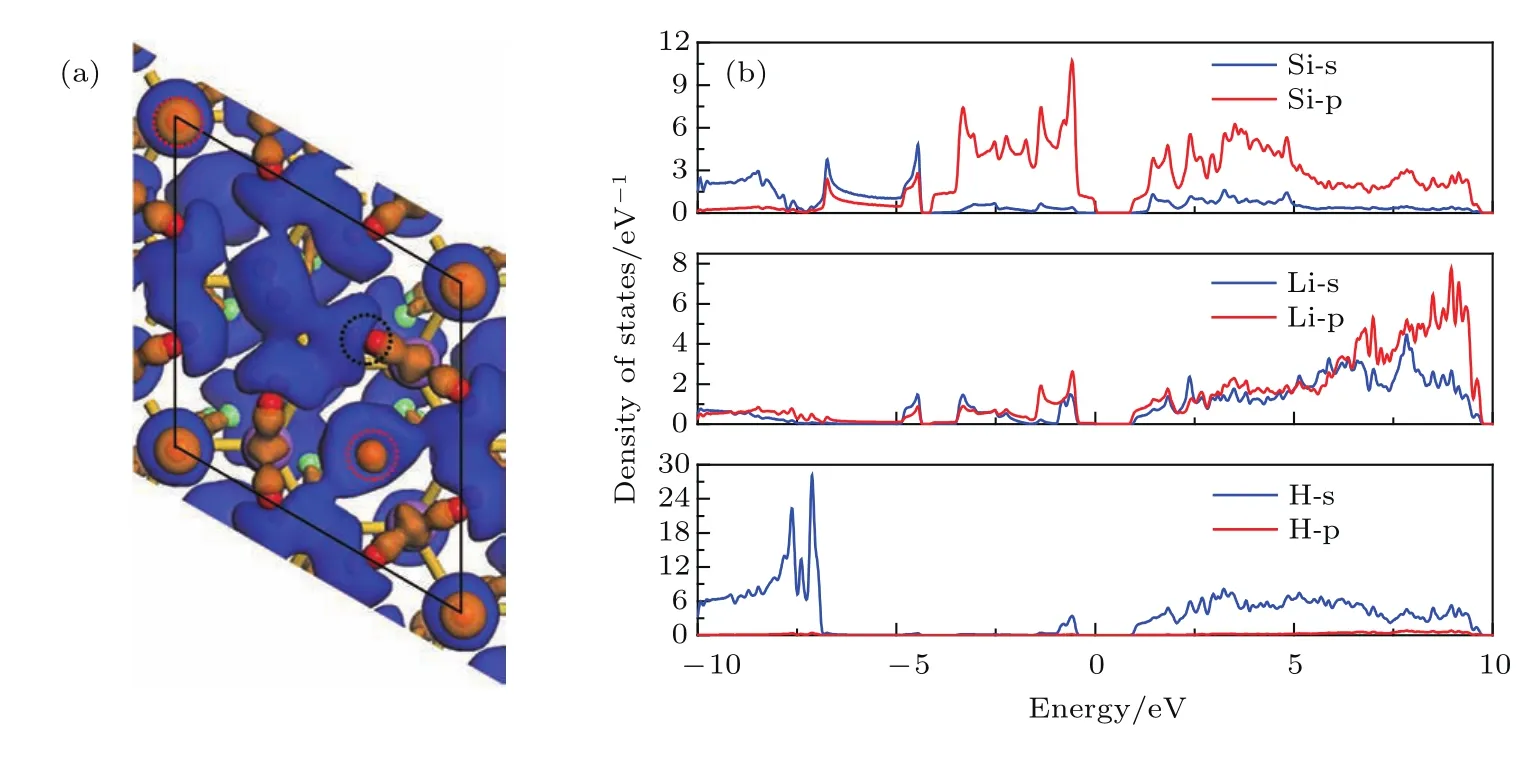
图7 Li0.50Si0.50-18H2硅烯结构的(a)差分电荷密度图,(b)态密度图Fig.7.Diagrams of(a)charge density differences and(b)density of states of Li0.50Si0.50-18H2.
此外,从图7(b)的态密度图中可以看到,H的s和p轨道同Li,Si的s和p轨道存在不同程度的杂化(−0.95—−0.44 eV以及0.98—9.65 eV),说明轨道杂化作用在Li修饰硅烯氢存储过程中也发挥了重要作用.结合Mulliken电荷布居的分析,发现各H原子从Li原子处获得了数量不等的电子,说明轨道杂化作用加强了Li正离子、Si负离子和极化氢气分子之间的静电作用.值得注意的是,存在两类氢气分子(图7(a)中红色虚圆内)周围的电荷密度增减情况与其他氢气分子不同,且其吸附距离相较于其他各氢气分子较大(2.37 Å和2.75 Å).本文尝试在结构优化前将这两类氢气分子远离硅烯表面,但在结构优化后这两类氢气分子仍然被吸附回硅烯表面,且其态密度图和其他氢气分子类似,因此我们认为上述两类氢气分子也被Li修饰硅烯所吸附,其吸附机理主要为轨道杂化作用.总体来看,氢气分子的氢键键长在吸附后略有增长(由0.748 Å变为0.766 Å),表明静电相互作用和轨道杂化作用只是促进了Li修饰硅烯对氢气的吸附,并未将氢气分子解离而破坏,这对于氢气分子在Li修饰硅烯中的存储和释放非常重要.
4 结 论
借助第一性原理计算方法,对不同Li吸附组分下硅烯的稳定性、氢存储性能和机理进行了系统性的研究.结果发现,在Li吸附组分达到0.50而饱和前,各硅烯体系中Li原子的平均结合能在大于其内聚能的基础上,随Li组分的增加而呈现增长趋势,表明在一定范围内提高Li修饰硅烯中的Li组分不会影响硅烯体系的稳定性,进一步说明本文研究不同Li组分下硅烯的氢存储性能具有可行性.在确保稳定性的基础上,通过依次添加氢气分子,发现硅烯体系的最大氢存储量会随着Li组分从0.11到0.50的增加而增大,并且当Li原子吸附达到饱和时达到最大,相应的最大质量储氢密度为11.46 wt%,平均吸附能为0.34 eV/H2,达到美国能源部制定的储氢标准,表明提高Li吸附组分至其饱和在理论上能有效提高硅烯的氢存储性能,使Li修饰硅烯成为良好的氢存储材料.同时发现,Li,Si离子与极化氢气分子之间的静电相互作用和H,Li,Si的s和p轨道之间的杂化作用是Li修饰硅烯可具备较大储氢能力的关键.上述研究结果揭示了Li修饰硅烯的储氢机理,并为充分挖掘Li修饰硅烯的氢存储性能和硅烯在未来氢存储领域的应用提供了理论指导.
[1]Cheng J Y,Chan M K Y,Lilley C M 2016Appl.Phys.Lett.109 133111
[2]Zhou J Q,Bournel A,Wang Y,Lin X Y,Zhang Y,Zhao W S 2017Appl.Phys.Lett.111 182408
[3]Yang S,Cheng P,Chen L,Wu K H 2017Acta Phys.Sin.66 216805(in Chinese)[杨硕,程鹏,陈岚,吴克辉2017物理学报66 216805]
[4]Hussain T,Kaewmaraya T,Chakraborty S,Ahuja R 2016J.Phys.Chem.C120 25256
[5]Li C,Yang S X,Li S S,Xia J B,Li J B 2013J.Phys.Chem.C117 483
[6]Li F,Zhang C W,Ji W X,Zhao M W 2015Phys.Status Solidi B252 2072
[7]Zhao J J,Liu H S,Yu Z M,Quhe R G,Zhou S,Wang Y Y,Liu C C,Zhong H X,Han N N,Lu J,Yao Y G,Wu K H 2016Prog.Mater.Sci.83 24
[8]Hussain T,Chakraborty S,De Sarkar A,Johansson B,Ahuja R 2014Appl.Phys.Lett.105 123903
[9]Wang Y S,Zheng R,Gao H Y,Zhang J,Xu B,Sun Q,Jia Y 2014Int.J.Hydrogen Energy39 14027
[10]Wang J,Li J B,Li S S,Liu Y 2013J.Appl.Phys.114 124309
[11]Ariharan A,Viswanathan B,Nandhakumar V 2017Graphene6 41
[12]Lochan R C,Head Gordon M 2006Phys.Chem.Chem.Phys.8 1357
[13]Song E H,Yoo S H,Kim J J,Lai S W,Jiang Q,Cho S O 2014Phys.Chem.Chem.Phys.16 23985
[14]Li F,Zhang C W,Luan H X,Wang P J 2013J.Nanopart.Res.15 1972
[15]Molle A,Grazianetti C,Cinquanta E 2016ECS Trans.75 703
[16]Zhong S Y,Ning F H,Rao F Y,Lei X L,Wu M S,Zhou L 2016Int.J.Mod.Phys.B30 1650176
[17]Hussain T,Kaewmaraya T,Chakraborty S,Ahuja R 2013Phys.Chem.Chem.Phys.15 18900
[18]Zhou C Y,Szpunar J A 2016ACS Appl.Mater.Interfaces8 25933
[19]Ma L,Zhang J M,Xu K W,Ji V 2015Physica E66 40
[20]Fair K M,Cui X Y,Li L,Shieh C C,Zheng R K,Liu Z W,Delley B,Ford M J,Ringer S P,Stampf lC 2013Phys.Rev.B87 014102
[21]Wang Y S,Li M,Wang F,Sun Q,Jia Y 2012Phys.Lett.A376 631
[22]Hussain T,Chakraborty S,Ahuja R 2013ChemPhys-Chem14 3463
[23]Delley B 2000J.Chem.Phys.113 7756
[24]Perdew J P,Chevary J A,Vosko S H,Jackson K A,Pederson M R,Singh D J,Fiolhais C 1993Phys.Rev.B48 4978
[25]Perdew J P,Wang Y 1992Phys.Rev.B45 13244
[26]Grimme S 2006J.Comput.Chem.27 1787
[27]Chadi D J 1977Phys.Rev.B16 1746
[28]Huang Y P,Yuan J M,Guo G,Mao Y L 2015Acta Phys.Sin.64 013101(in Chinese)[黄艳平,袁健美,郭刚,毛宇亮2015物理学报64 013101]
[29]Tritsaris G A,Kaxiras E,Meng S,Wang E G 2013Nano Lett.13 2258
[30]Liu C S,Zeng Z 2010Appl.Phys.Lett.96 123101
猜你喜欢
石油石化绿色低碳(2022年4期)2023-01-06
大学物理(2022年9期)2022-09-28
中国特种设备安全(2022年4期)2022-07-08
中国特种设备安全(2022年4期)2022-07-08
压力容器(2020年9期)2020-10-23
物理通报(2020年7期)2020-07-01
信息记录材料(2016年4期)2016-03-11
原子与分子物理学报(2015年3期)2015-11-24
医学研究杂志(2015年12期)2015-06-10
科学启蒙(2013年2期)2013-07-11
