
采用气相色谱法建立氟[18F]脱氧葡糖注射液中有机溶剂残留的测定方法及其方法不确定度的评价
2018-06-14 06:44:44陈立光梁明泉邓庆荣
中国医药科学 2018年9期
陈立光 梁明泉 邓庆荣 罗 文
1.深圳市保健委员会办公室PET/CT诊断中心,广东深圳 518020;2.中国广州分析测试中心,广东广州 510070
PET/CT显像已成为核医学发展的新“亮点”,18F标记的2-去氧-2-18F氟-D-葡萄糖注射液(18F-FDG)被认为是本世纪很有价值的放射性药物。18F-FDG属于正电子类放射性诊断用药,目前临床广泛应用于PET/CT肿瘤诊断中,经过在国内近20年的临床试用,《中国药典(二部)》(2015版)开始将18F-FDG收载为氟[18F]脱氧葡糖注射液[1]。随着我国PET/CT扫描仪普及以及回旋加速器药物生产系统投入使用,氟[18F]脱氧葡糖注射液的制备、药品质量控制和用药安全越来越受到国家药监部门的重视。氟[18F]脱氧葡糖注射液是当前临床上正电子发射断层显像(PET)技术最常用的示踪剂[2-4],18F-FDG在PET中的用量占到了95%以上[5]。18F-FDG制备工艺多通过计算机控制的自动化有机合成和纯化制备,而在制备工艺中多采用有机溶剂乙腈、乙醇等[1,6],最终产品的有机溶剂残留不可避免,因此对有机溶剂残留的质量控制尤为重要。按照2015年版《中国药典》对残留溶剂的限度要求[7],第二类溶剂中乙腈的残留限度为0.041%,第三类溶剂乙醇的限度为0.50%。本研究主要探究气相色谱分析法对18F-FDG产品的有机溶剂残留进行分析。建立一种对氟[18F]脱氧葡糖注射液中乙腈、乙醇两种有机溶剂的残留量进行快速、定量测定的方法,用于日常生产中对氟[18F]脱氧葡糖注射液的质量控制,具体报告如下。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器设备 Eclipse RD 型Cyclotron 回旋加速器(西门子公司);药物生产系统(专利研发,发明专利申请号:20171068609.4);BBS1 38B1B/1型合成防护防护热室,COMECER 公司产品;CRC-15R 型活度计(美国CAPINTEC公司生产);Agilent 6820型气相色谱仪:Agilent公司,氢火焰离子化检测器(FID)氮磷检测器(NPD)检测器,QA/QC Cerity工作站,毛细管柱SUPELCOSPB-5(30m×0.25mm×0.25um); 精 密 电 子 天 平(Sartorius公司);SW-CJ-1D型净化工作台,苏州净化设备有限公司。
1.1.2 试剂 H218O:丰度95%(ABX公司产品);2-三氟甲基磺酰基-F-D甘露糖(三氟甘露糖),Kryptofix2.2.2(K2.2.2),无水乙腈、无水乙醇(UPS级)(均 为 Sigma-Aldrich公 司);QMA柱,Alumin-N柱,C18柱(Waters公司);AG50树脂和AG11A8树脂:50 ~ 100目(Bio-Rad公司出品);HPLC级去离子水,自备;压缩空气,高纯氢气及氮气(深圳联华气体有限公司产品)。
2 实验方法
2.1 氟[18F]脱氧葡糖注射液的制备
氟[18F]脱氧葡糖注射液的制备包括标准的六个步骤。(1)首先是18F-负离子的产生。从18O富氧水开始,通过p-n核反应,18O原子核俘获一个质子的同时发射一个中子而产生18F-负离子。这一步依赖于回旋加速器;(2)俘获和释放18F-负离子。这一步基于离子交换的原理;(3)反应活性18F-负离子的制备。这一步的主要目的是通过乙腈和水的共沸原理移除反应体系中的水分,保证第4步的标记反应体系严格要求在疏水的溶液体系中进行;(4)反应活性放射性18F-负离子和 1,3,4,6-四-O-乙酰基-2-O-三氟甲磺酰-β-d-吡 喃 甘 露 糖(1,3,4,6-Tetra-O-acetyl-2-O-trifluoromethanesulfonyl-β-D-mannopyranose)发生亲核反应,产生18F-负离子辐射标记的甘露糖三氟磺酸酯;(5)水解移除乙酰基保护基团。(6)纯化产物,这一步移除各种中间产物并作无菌、无热源等纯化处理,使用0.22µm滤膜过滤得到氟[18F]脱氧葡糖注射液,并将其收集于无菌真空瓶中。
2.2 气相分析条件
分离柱:SUPELCO SPB-5型毛细管柱,高纯氮气压力设置为50kPa,空气压力设置为50kPa,高纯氢气压力设置为100kPa,SPB-5毛细管分离柱的温度设置为60℃,FID监测器温度设置为150℃,气化室温度设置为150℃。高纯氢气20mL/min,空气300mL/min,和高纯氮气10mL/min;检测后得到标准峰面积,并以峰面积为纵坐标,有机溶剂浓度为横坐标,进行线性回归分析。乙腈和乙醇的标准溶液的制备采用相同方法,并绘制各自的线性工作曲线。
2.3 工作曲线的建立
使用精密电子天平称取200.00mg乙腈,将其放置于50mL容量瓶中,加水定容,摇匀,得到标准溶液,浓度为4.000g/L。使用移液管移取上述标准品 10.0、5.0、2.5、0.5mL 至 20mL 容量瓶中,加水定容,摇匀,分别得到浓度为 2.00、1.00、0.50、0.10g/L溶液,然后按照2.2的气相条件分别进行气相色谱检测, 进样量为2µL,检测后得到标准峰面积,以标准品的浓度作为横坐标,以标准峰面积作为纵坐标,计算回归方程及线性及工作曲线;每个样品进样两次,取平均值。
2.4 系统回收率与重复性的测定
根据2.3中建立的方法,配制乙腈和乙醇有机溶液,取浓度为0.80g/L的乙腈和乙醇标准有机溶液,进行气相分析,测定5次,且每次进样量相同,计算平均值,并使用标准曲线方程计算浓度,得出回收率。再取浓度为0.80g/L的乙腈和乙醇标准有机溶液,相同条件下测量10次,检测系统重复性。
2.5 18F-FDG有机溶剂残留的气相色谱法检测
按照2.2的分析条件,分别对随机抽取5批次的氟[18F]脱氧葡糖注射液进行气相色谱检测,根据标准曲线方程计算得到氟[18F]脱氧葡糖注射液中乙腈和乙醇的有机残留量。
3 结果与讨论
3.1 标准样品有机溶剂的线性关系
通过对两种有机残留溶剂含量的气相分析,测定其峰面积。绘制线性工作曲线,其中横坐标是标准溶液浓度,纵坐标则为峰面积。得到其曲 线方 程分 别 为:y=2287.6X-47.644,r=0.9994;y=1366.5X+85.821,r=0.9995; 线性范围为0.10~2.00g/L。该结果说明两种溶剂的峰面积与浓度的线性关系良好。
3.2 标准有机溶剂气相检测
两种标准品溶液的气相色谱图见图1,分析时间为15min。按照保留时间顺序依次为乙醇、乙腈,其中乙醇的保留时间为1.84min;乙腈依次为的保留时间为2.04min;两种溶剂的检测均可在10min内完成。信噪比按照3倍来计算,可测的最低检测浓度为0.031g/L,以进样量为2µL进行计算,在该检测分离条件下,乙腈和乙醇的最低检测线分别为0.22µg、0.20µg。

图1 乙腈和乙醇的标准色谱圈
3.3 系统回收率和精密度
5次测量的平均值及回收率的测量通过标准曲线方程进行计算,经计算,得出乙醇和乙腈的平均值分别为0.078、0.079g/L,回收率分别为98.7%、98.4%。平均值及回收率结果说明系统具有良好的准确性。采用该系统条件对10批次样品进行检测,结果显示,乙腈和乙醇的含量 分 别 为(0.078±0.02),(0.079±0.03)g/L,说明系统测量重复性良好,精密度可以满足检测要求。
3.4 样品分析
随机抽取5批次制备的氟[18F]脱氧葡糖注射液样品,按照2.3气相条件进行检测,两种残留溶剂的检测结果见表1。由表1可见虽然不同批次中残留溶剂含量不同,但是均未超过《中国药典》2015 年版四部对氟[18F]脱氧葡糖注射液的质量控制要求规定的含量(第二类溶剂中乙腈的残留限度为0.041%,第三类溶剂乙醇的限度为0.50%。)[7],同时也符合美国药典的标准[8],因此可以认为该药品中的残留溶剂含量标准相同均为0.50%。
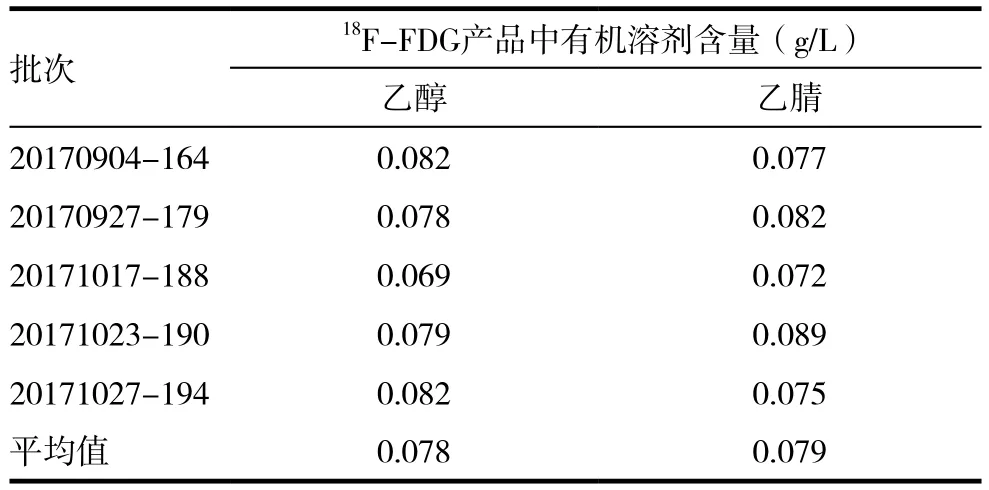
表1 5批次氟[18F]脱氧葡糖注射液中有机残留乙醇、乙腈的含量
3.5 氟[18F]脱氧葡糖注射液中有机溶剂残留的测定方法的不确定度
(1)不确定度的来源分析。本研究所采用的分析方法的不确定度来源主要有:①色谱柱;②气化室和检测器;③乙腈浓度;④人员操作;⑤仪器本身的稳定性。
(2)不确定度分析。①色谱柱:在实验过程中发现,色谱柱的柱温、柱效均会对两种物质的分离效果产生影响,而不同类型的柱子直接关系到该两种有机物质能否有效分离。有研究指出,乙腈、在极性较小的柱子上较难分离,出峰时间无法分开[9-10]。②气化室和检测器:气化室的温度不宜过高,当其温度高于120℃时,会使待测样品的保留时间缩短[11],但是不会对样品的检测造成影响。③乙腈浓度引起的不确定度:经验表明:在制备氟[18F]脱氧葡糖注射液的过程中,乙腈起到很重要的作用,主要用作反应前体以及催化剂穴醚的溶剂[12-13],而大部分作为载体的乙腈会在和水的共沸蒸发、亲和反应和后续的高温蒸发过程被除掉,因此,反应过程中乙腈的最终浓度可以对整个反应起到一定的反应性。④人员操作:不同工作人员的工作态度不同,且操作以及称量样品的习惯均会有所差异[14-15],因此尽量由一位工作人员完成。⑤仪器本身的稳定性引起的不确定度:由于气相过于灵敏,因此极易出现损坏,所以在使用前需要校准[16]。
4 结论
乙腈和乙醇溶剂是制备氟[18F]脱氧葡糖注射液过程中无法避免的有机残留物,乙腈被美国药典规定为二类限制使用溶剂,乙醇在美国药典中为三类溶剂,且均对其残留量进行了限定;我国《中国药典》2015年版四部对氟[18F]脱氧葡糖注射液的质量控制要求规定的含量,该药品残留溶剂测定法采用通则0861第一法进行试验。在气相色谱检测中,如果检测器为带氢火焰检测器(FID),则乙腈和乙醇的残留量应分别低于0.04%和0.5%,而且中国药典中规定气相检测须在药品使用前完成。本次检测结果显示,虽然氟[18F]脱氧葡糖注射液的终产品中乙腈和乙醇的含量高低不同,但均没有超出《中国药典》2015 年版四部对氟[18F]脱氧葡糖注射液的质量控制要求,证明了合成药品的质量完全符合临床用药标准。另外采用气相色谱法进行检测,可以准确的对氟[18F]脱氧葡糖注射液的中的有机残留量进行测定,而且检测时间仅为10min。该检测方法简单迅速,尤其适用于半衰期短的PET示踪剂药物,为临床用药安全提供了保障。
[1] 李军.新型18F-脱氧葡萄注射液的制备与质量控制[J].华西药学杂志,2004,19(1):8-10.
[2] 张锦明.正电子放射性药物进展[J].中华核医学杂志,2003,23(5):315-317.
[3] 刘芳蕾,洪群英,石洪成,等.18氟-氟代脱氧葡萄糖正电子发射计算机断层扫描在肺癌早期诊断中的应用价值 [J].中华医学杂志,2013,93(38):3019-3022.
[4] 张悦,张遵城.双时相18F-FDG PET显像的应用进展 [J].山东医药,2015,55(39):101-103.
[5] 范文博.自动化合成2-18F-β-D-脱氧葡萄糖及其质量控制 [J].化工时刊,2014,28(6):1-3.
[6] 何山震,王淑侠,陈立光,等.气相色谱法对18F-FDG中有机溶剂残留的测定[J].同位素,2008,21(1):58-60,64.
[7] 中华人民共和国药典二部:附录124-125[M].北京:中国医药科技出版社,2015.
[8] Iaea.Iaea safeguards Glossary[M].Vienna:lAEA,2002:33.
[9] DouG Reilly.Norbert Ensslin,Hastingssmith.et a1.Passive Nondestructive Assay of Nuclear Mateals:LA-uG90-732[M].NewMexico:LANL,199l:Vii.
[10] Morel J,Etcheverry M,Riazuelo G.Uranium Enrichment Measurement by X-ray Spectrometry With the Process[J].Applied Radiation and Isotopes,1998,49(9):1251.
[11] 宋晓勇.气相色谱仪操作、使用、检定校准方法[J].计量与测试技术,2013,40(6):48..
[12] 花宁.气相色谱法直接测量,18F-FDG中Kryptofix2.2.2 的含量 [J].同位素,2007,20(2):105-107.
[13] 李云钢,张晓军,刘健,等.气相色谱法测量正电子放射性药物中有机溶剂残留及原因分析[J].同位素,2013,26(3):152-157.
[14] 唐安戊,劳海燕,刘冰,等.动态比浊法鲎试验定量测定18F-FDG的细菌内毒素含量[J].中华核医学杂志,200l,21(4):249-250.
[15] 邓鹏裔.住友F300E和国产FDG-N模块的18F-FDG产品质量对比研究[C].//2015全国同位素制备及应用技术交流研讨会论文集.2015:80.
[16] 于乃海,彭川荣,孙德政,等.气相色谱仪校准测量结果不确定度的评定[J].山东电力技术,2006,5(2):22-25.
猜你喜欢
中国药物经济学(2023年12期)2023-02-02 07:36:26
现代医药卫生(2021年23期)2021-12-05 19:04:11
食品与生物技术学报(2021年4期)2021-01-17 04:06:35
家庭医学(下半月)(2020年7期)2020-04-18 13:45:31
北方牧业(2016年1期)2016-12-17 19:08:50
癌变·畸变·突变(2016年4期)2016-08-22 05:52:34
中国医药生物技术(2014年4期)2014-01-23 09:24:24
中国信息化·学术版(2013年6期)2013-09-30 06:39:40
食品科学(2013年23期)2013-03-11 18:29:49
河北大学学报(自然科学版)(2012年3期)2012-03-25 10:13:01
