
高熔体弹性PLA/咖啡渣复合材料的发泡行为研究
2018-06-05 04:17:37何仕成杨余杰周洪福
中国塑料 2018年5期
刘 伟,何仕成,杨余杰,王 跃,周洪福
(1.贵州理工学院材料与冶金工程学院,贵阳 550003;2.北京工商大学,中国轻工业绿色塑料成型技术与质量评价重点实验室,北京 100048)
0 前言
PLA是一种代表性的可降解高分子材料,具有生物相容性、良好的物理和力学性能[1-2]。咖啡渣是生活中常见的完全生物降解天然高分子生物质[3]。目前,这种生物质资源大量通过堆肥、填埋或焚烧等方式处理,资源综合化利用程度极低[4]。上述2种材料通过共混后可以成型完全生物降解制品,还能够大大降低PLA原材料成本。进一步发泡成型后有应用于包装或生物医药等多个领域的潜在价值和环保意义。然而,现在普遍认为PLA本身熔体弹性低,与咖啡渣共混还将继续降低其熔体弹性,难以成型有使用价值的发泡材料[5-6]。因此,提高熔体弹性对于制备发泡材料是关键因素。解决该问题最为有效的方法是扩链法[7]。MDI是一种高反应活性的扩链剂。首先,MDI的异氰酸基与PLA分子链的端羟基和端羧基进行反应具有扩链效果;其次,MDI与咖啡渣表面的羟基反应产生桥接作用,改善复合材料的界面结合力。
目前,高熔体弹性PLA/咖啡渣复合材料的相关研究较少,特别是利用MDI改性PLA/咖啡渣复合材料的发泡行为研究更是鲜有报道。因此,本文利用反应法制备PLA/咖啡渣复合材料,研究PLA/咖啡渣复合材料的熔体弹性等流变性能、诱导结晶性能和发泡行为的关系。研究工作的开展对于确定合理的工艺条件、对于探索咖啡渣在PLA发泡材料中的综合应用,以及制备高性能可降解发泡材料具有一定意义。
1 实验部分
1.1 主要原料
PLA,4032D,美国Natureworks公司;
咖啡渣,粒径约为5 μm,美国星巴克公司;
MDI,分析纯,国药集团化学试剂有限公司。
1.2 主要设备及仪器
密炼机,XSS-60,上海科创橡塑机械设备有限公司;
超临界CO2固相发泡设备,自制;
精密鼓风干燥箱,BPG-9140A,上海一恒科学仪器有限公司;
差示扫描量热仪(DSC),Q100,美国TA仪器公司;
高级动态流变仪,MARS,美国赛默飞世尔科技哈克公司;
力学万能试验机,CMT6104,新三思计量技术有限公司;
数显冲击试验机,XJZ-50,承德试验机有限责任公司;
扫描电子显微镜(SEM),Nova Nano 450,美国FEI公司;
样品表面喷金仪器,EMITECH K550X,捷克泰思肯仪器公司;
密度天平,JA3003J,上海恒平仪器公司。
1.3 样品制备
PLA/咖啡渣复合材料制备:利用鼓风干燥箱在60 ℃温度下干燥12 h去除PLA原料表面水份;收集的咖啡渣首先利用酒精清洗并使用鼓风干燥箱在60 ℃温度下干燥12 h;使用密炼机熔融共混制备PLA/咖啡渣、MDI改性PLA和PLA/咖啡渣复合体系,熔融共混温度为190 ℃,密炼时间为6 min,转速为60 r/min,共混配比如表1所示,熔融共混后的样品进一步干燥处理(干燥条件同上),提供流变性能、结晶性能和力学性能测试样品和进一步制备发泡样品;
PLA/咖啡渣发泡材料制备:将不同配比的PLA/咖啡渣复合材料置于高压釜内,升温至150 ℃,将CO2注入到高压釜内,并维持至20 MPa,使PLA/咖啡渣复合材料浸泡在超临界CO2中4 h,CO2分子充分溶解于PLA基体中并达到平衡。随后,分别降温至低于冷结晶温度(120 ℃)和冷结晶温度(130 ℃)下并释放釜内压力至常压,得到不同配比的PLA/咖啡渣复合发泡材料,截取发泡芯层作测试样品。
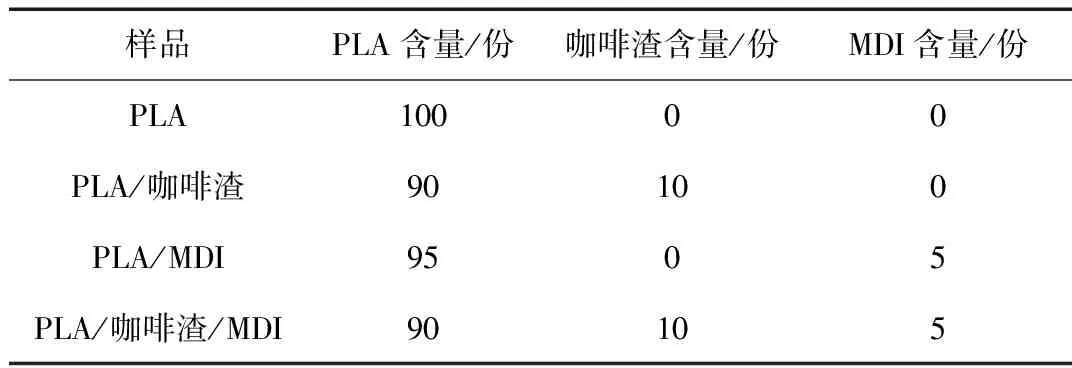
表1 试验配方表Tab.1 Experimental formula
1.4 性能测试与结构表征
熔体弹性表征:利用高级动态流变仪对PLA/咖啡渣复合材料的动态剪切流变数据进行表征,试验样品置于直径为20 mm的圆形平行板间,测试间距为1 mm,测试温度为190 ℃,频率范围为0.1~100 s-1;
热性能表征:利用DSC对PLA/咖啡渣复合材料的结晶和熔融行为进行表征;测试时样品处于氮气氛围,以10 ℃/min的速率加热至190 ℃,观察其冷结晶和熔融行为,PLA/咖啡渣复合材料的结晶度按照式(1)计算:
(1)
式中Xc——PLA的熔融结晶度,%
ΔHm——样品熔融焓,J/g
ΔHcc——样品的冷结晶焓,J/g
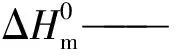
力学性能表征:拉伸性能按GB/T 1040.1—2006进行测试,拉伸速率为5 mm/min,每个配方至少测试5根样条,并取平均值;冲击强度性能按GB/T 1843—2008进行测试,测试使用带V形缺口的简支梁冲击方式,冲击能量2 J,每个配方至少测试5根样条,并取平均值;
分散结构和泡孔结构表征:PLA/咖啡渣复合材料发泡材料的密度采用排水法进行测试,每组样品测试3次,取平均值作为该样品的密度;采用SEM对PLA/咖啡渣复合体系的分散结构和泡孔结构进行表征;样品浸泡在液氮中完全冷却并脆断,断面表面喷金,观察分散结构和泡孔结构;泡孔尺寸利用Image Tool软件统计,泡孔密度通过式(2)进行计算:
(2)
式中Nc——泡孔密度,个/cm3
ER——发泡材料的发泡倍率
R——扫描电镜照片中发泡材料的泡孔平均直径,μm
2 结果与讨论
2.1 PLA/咖啡渣复合材料的分散形态
■—PLA ●—PLA/咖啡渣 ▲—PLA/MDI ▼—PLA/咖啡渣/MDI(a)G′-ω曲线 (b)tanδ-ω曲线 (c)η*-ω曲线 (d)Cole-Cole曲线图2 PLA/咖啡渣复合材料的流变曲线Fig.2 Rheological curves of the PLA/coffee ground composites
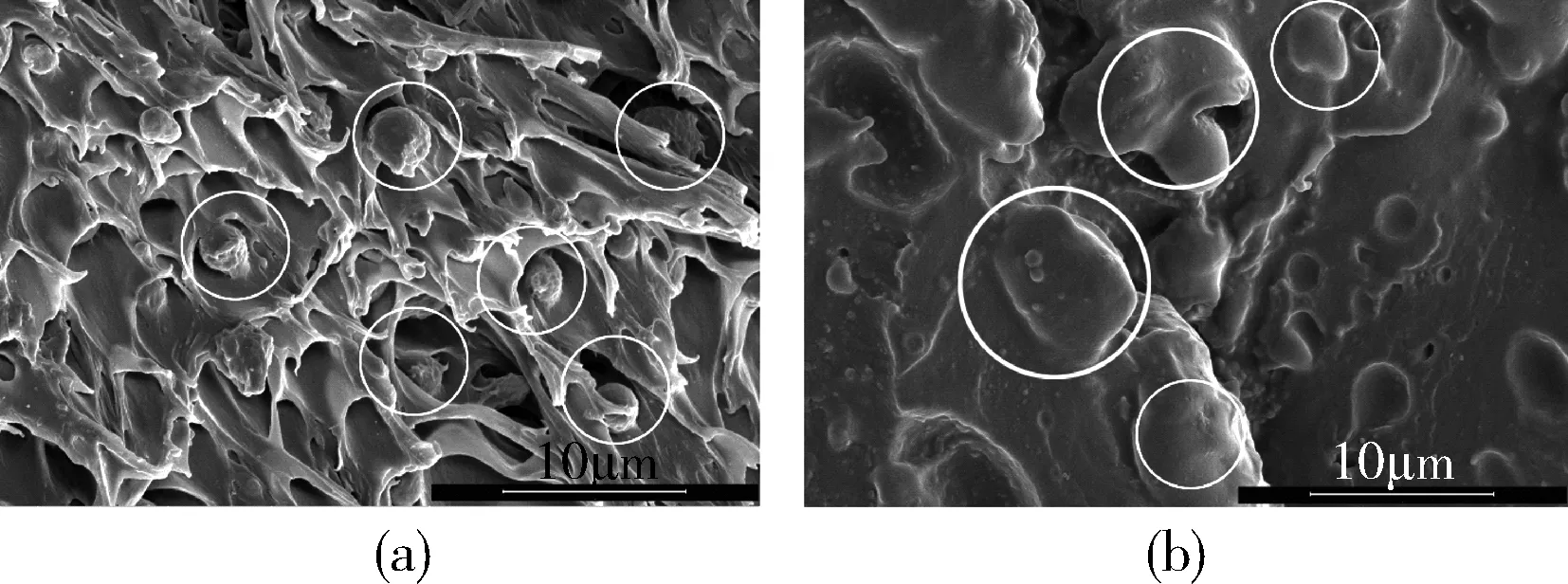
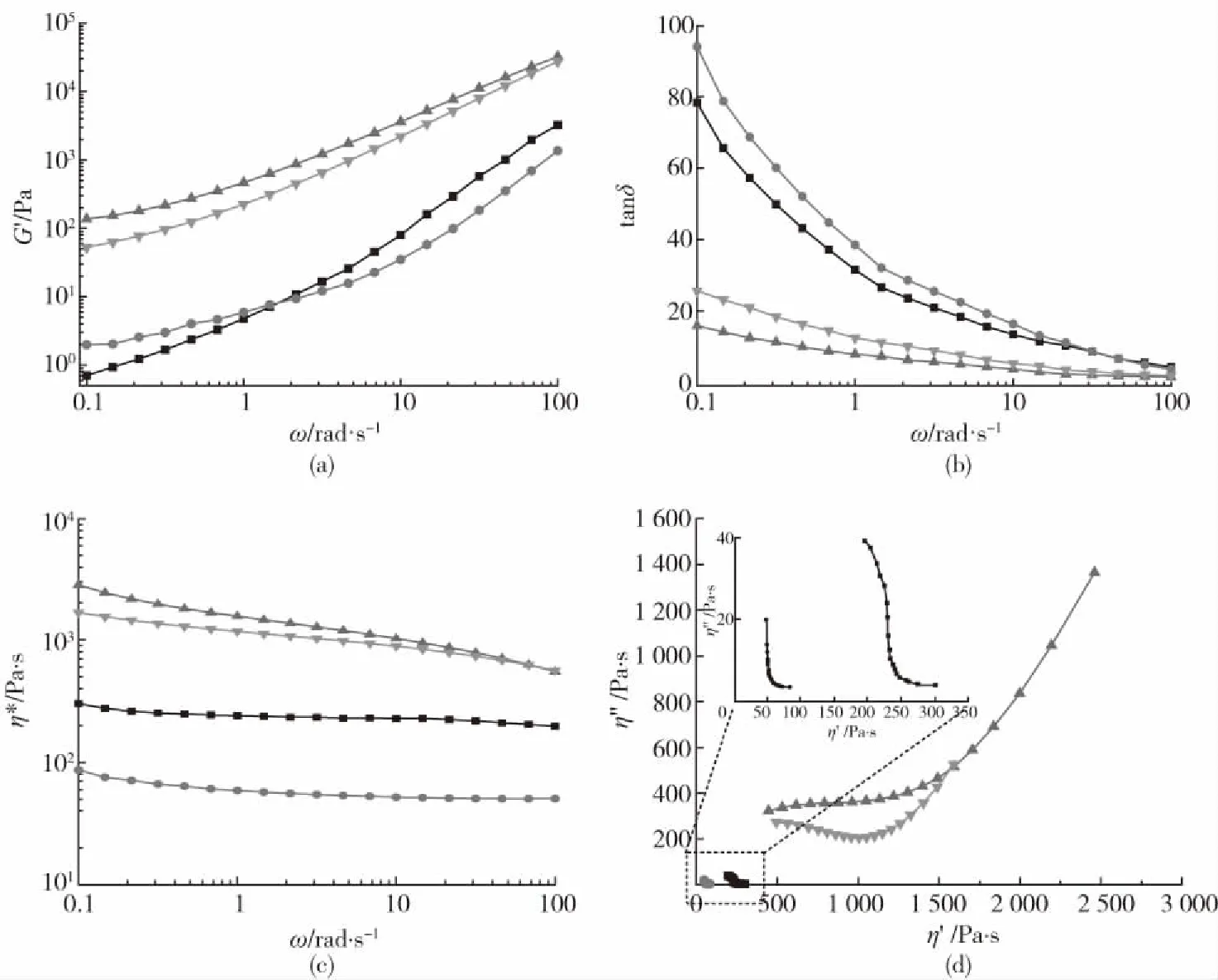
样品:(a)PLA/咖啡渣 (b)PLA/咖啡渣/MDI图1 PLA/咖啡渣复合材料分散形态的SEM照片Fig.1 SEM photos of the PLA/coffee ground composites
分散相在基体中的分散形态和界面结合作用是影响复合材料性能的关键因素。图1为PLA/咖啡渣复合材料和使用MDI改性PLA/咖啡渣复合材料的分散形态。从图中可以看出,通过熔融法直接将PLA和咖啡渣共混后,分散相咖啡渣在PLA基体相中呈球形分布,分散相的尺寸约为5 μm,且分散相与基体相的界面清晰,彼此脱离。这种现象说明咖啡渣与PLA的相容性差,界面结合力弱,分散相易于与基体相发生分离。对比可见,利用MDI改性PLA/咖啡渣复合材料中两相界面结合力明显提高,较少观察到脱离基体的咖啡渣分散相,咖啡渣被基体树脂所包裹。这种现象主要是MDI的异氰酸基和咖啡渣表面的羟基以及基体树脂反应有关。MDI起到良好的桥接作用,使PLA能够包裹分散相,较好提高界面结合力。
2.2 PLA/咖啡渣复合材料的流变性能
PLA/咖啡渣复合材料的发泡行为与熔体弹性密切相关,而熔体弹性能使用动态流变性能进行直接表征。图2为PLA/咖啡渣复合材料的储能模量(G′)、损耗因子(tanδ)、复数黏度(η*)和Cole-Cole曲线。如图2(a)所示,储能模量是PLA/咖啡渣复合材料的熔体黏弹性的弹性大小。一般而言,储能模量值越大,熔体弹性越明显。从图2(a)中可以看出PLA和PLA/咖啡渣复合材料的储能模量均表现出明显频率(ω)依赖性,且纯PLA的储能模量值低,说明其熔体弹性弱。加入咖啡渣后,PLA的储能模量即熔体弹性变化不大,体系仍以PLA的熔体弹性特征为主导。利用MDI对PLA和PLA/咖啡渣复合体系改性后,2种体系的的储能模量提高约2个数量级。这种现象说明了MDI的异氰酸基团与PLA分子链端基和咖啡渣表面的羟基的充分反应,产生扩链效果和桥接作用。这种分子链结构的变化导致其储能模量偏离经典的线性黏弹理论,即G′∝ω2,从而出现“第二平台”现象,在加入MDI的体系中均能够观察到这种典型的末端区效应,表明加入MDI有利于制备具有高熔体弹性的PLA/咖啡渣复合材料。
图2(b)是PLA/咖啡渣复合材料的损耗因子曲线。损耗因子是PLA/咖啡渣复合材料在交变剪切应力下,应变和应力相位差的正切值,也是衡量熔体弹性和黏性的贡献值。通常其值越小,说明熔体弹性贡献越大,复合材料的可发性越好。从图2(b)中能够观察到PLA在低频区内,其熔体弹性响应低于黏性响应。加入咖啡渣后,复合体系的黏性响应进一步提高。熔体黏性贡献对可发性影响较小,可发性主要受控于熔体弹性贡献。在2种体系中加入MDI后,可以看出末端区的损耗因子显著降低,熔体弹性贡献大于熔体黏性贡献部分。相比于PLA/咖啡渣复合体系,MDI改性的PLA/咖啡渣复合体系的熔体弹性贡献明显高。这种特性使得复合材料在受交变应力作用时也比较稳定,有利于在发泡过程中维持泡孔结构。
图2(c)为PLA/咖啡渣复合材料的复数黏度曲线。从图中可以看出复数黏度随着频率增加而降低,是典型的剪切变稀的现象。在PLA基体中,随着咖啡渣加入,复合材料的复数黏度降低。产生这种现象可能与咖啡渣在加工过程中促进PLA热降解有关,Najafi等研究发现向PLA中加入含有羟基的填料时容易造成热降解使相对分子质量下降[9],而相对分子质量降低则会导致复数黏度下降。加入MDI后,不论是PLA或PLA/咖啡渣复合体系,其基体树脂相对分子质量提高,分子链的热运动和链间相互穿梭的运动受到限制,从而表现为复数黏度提高。复数黏度是控制发泡过程中泡孔结构固化和稳定的关键因素,当泡孔形成后,过低的复数黏度易于导致泡孔在固化时产生泡孔合并现象。所以,一定程度提高复数黏度有利于稳定泡体结构。
依赖于储能模量或复数黏度的熔体弹性常常表现为瞬态值,为了更准确研究PLA/咖啡渣复合材料黏弹性,确定MDI对复合材料的末端松弛时间变化,利用实数黏度η′与虚数黏度η″作图得到Cole-Cole曲线可以表征末端松弛时间。图2(d)是PLA/咖啡渣复合材料的Cole-Cole曲线。从图中能够观察出曲线呈半圆形。而曲线对应的直径与末端松弛时间相关。MDI改性后PLA分子链变长,同时咖啡渣存在一定的表面接枝链,更容易形成分子链缠结。这种缠结类似物理交联点,在分子链热运动过程中会延长其末端松弛时间。所以MDI改性的PLA及其复合体系曲线一定程度偏离半圆形,在曲线的末端有上扬的趋势。一般认为上扬的趋势与缠结程度有关:上扬程度越大,说明分子链的缠结程度越高,末端松弛时间越长。较长的末端松弛时间是高熔体强度PLA的典型流变表现,有利于在泡孔生长阶段维持泡孔壁的稳定,减少泡孔破裂现象。
2.3 PLA/咖啡渣复合材料的热性能
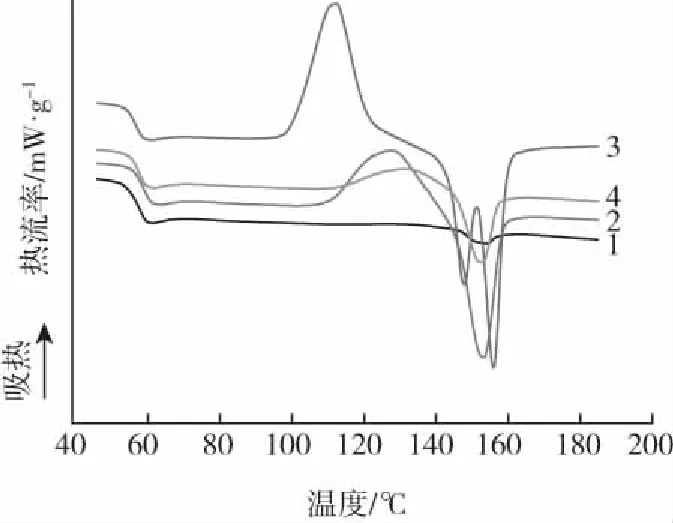
样品:1—PLA 2—PLA/咖啡渣 3—PLA/MDI 4—PLA/咖啡渣/MDI图3 PLA/咖啡渣复合材料的DSC曲线Fig.3 DSC curves of the PLA/coffee ground composites
图3是PLA、PLA/咖啡渣复合材料和对应MDI改性样品的熔融升温曲线图,表2为熔融曲线中对应的冷结晶峰温(Tcc)、熔融峰温(Tm)、熔融焓值和结晶度等参数。从图2可以看出,由于PLA分子链刚性较大,所以其加热过程中几乎没有吸热峰或放热峰,即没有明显的冷结晶和熔融热行为,且结晶度仅为1.22 %。当向PLA中加入咖啡渣后,熔融曲线中出现一个明显的冷结晶峰和一个熔融峰,对应的峰温分别为126.98 ℃和152.79 ℃。这种现象的产生可能与咖啡渣造成PLA的降解有关:PLA的相对分子质量降低,短链运动能力强,结晶度提高至4.71 %。然而,利用MDI对PLA进行扩链改性后,PLA表现出一个尖且窄的冷结晶峰和熔融双峰,熔融双峰温度在156 ℃和148 ℃左右。有文献报道出现熔融双峰与MDI扩链反应诱导结晶和冷结晶相关[10]。相比PLA/咖啡渣复合材料,MDI改性PLA的冷结晶峰由12.44 J/g大幅提高至23.10 J/g。熔融双峰中的低温峰与冷结晶相关,而高温峰与降温过程的结晶相关。对于MDI改性的PLA/咖啡渣复合材料冷结晶峰和熔融峰较弱,原因可能与PLA、咖啡渣和MDI之间的相互反应有关。结晶性能变化对PLA/咖啡渣复合材料可发性具有调控作用。其一是在发泡过程中,CO2仅在无定型区域内溶解和扩散,可发性受控于结晶度。其二是发泡过程中诱导的大量结晶可以作为泡孔成核点[11]。2种效应有利于改善发泡材料的泡孔尺寸、发泡倍率和泡孔形态。

表2 PLA/咖啡渣复合材料的热性能参数Tab.2 Thermal property parameters of the PLA/coffee ground composites
注:Tg——玻璃化转变温度,℃。
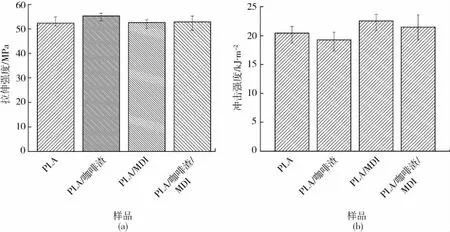
(a)拉伸性能 (b)冲击性能图4 PLA/咖啡渣复合材料的力学性能Fig.4 Mechanical properties of the PLA/coffee ground composites
2.4 PLA/咖啡渣复合材料的力学性能
图4为PLA/咖啡渣复合材料的力学性能。从图4(a)可以看出PLA的拉伸强度为52 MPa,是一种典型的刚性材料。加入咖啡渣后,复合材料的拉伸强度增加至55 MPa。这种增强效应主要归因于咖啡渣的诱导结晶效应,低相对分子质量的PLA易于冷却结晶,而高结晶度有利于增强复合体系[12]。相反,加入MDI后复合材料的拉伸强度降低至52 MPa,其原因可能是MDI改变PLA分子链的规整性,降低冷却结晶能力,使得复合材料的拉伸强度出现下降趋势。图4(b)为复合材料的冲击强度,PLA的冲击强度为20 kJ/m2,加入咖啡渣后降低至19 kJ/m2。随后加入MDI,复合材料的冲击强度反而提高至22 kJ/m2。冲击强度与拉伸强度的变化趋势相反,说明复合材料的力学性能主要受控于结晶诱导作用,较高的结晶度有利于增强,反之利用MDI的扩链作用破坏分子链的规整度降低结晶度有利于增韧。
2.5 PLA/咖啡渣复合材料的发泡性能
因为PLA/咖啡渣复合材料具有冷结晶现象,选择在130 ℃下诱导冷结晶并进行发泡,同时选择120 ℃以对比没有冷结晶时对发泡的影响。图5为不同发泡温度的PLA/咖啡渣复合材料的泡孔形态图。从图5中可以看出在2个不同的发泡温度下,PLA的泡孔结构均匀性很差,泡孔为不规则状,说明泡孔在生长过程中经历了大量的合并和破裂。加入咖啡渣后,复合材料的泡孔结构均匀性进一步变差,这与流变行为研究结果一致。然而,利用MDI改性后,PLA和PLA/咖啡渣复合材料的泡孔合并和破裂的现象大幅减弱,球形或五边十二面体泡孔结构较为清晰。复合材料的泡孔形态的改善与熔体弹性和末端松弛时间提高有关,从而泡孔形态得到改善。表3为不同发泡温度下PLA/咖啡渣复合材料的泡孔结构参数。一方面,对于没有MDI改性的样品,在发泡温度为120 ℃时,由于PLA和PLA/咖啡渣复合材料的熔体弹性差,2种样品发泡后发泡倍率仅为4.34倍和3.58倍,泡孔密度只达到3.74×106个/cm3和2.63×106个/cm3。相比之下,在发泡温度为130 ℃时,样品的发泡倍率和泡孔密度均有小幅度提升,这种现象与诱导冷结晶程度有关。另一方面,对于MDI改性的样品,PLA/咖啡渣复合材料的熔体弹性大幅改善,其发泡倍率由3.58倍提高至7.24倍。此外,泡孔密度也由2.63×106个/cm3提高至1.79×107个/cm3。此外,进一步将发泡温度由120 ℃提高至130 ℃,即处于PLA/咖啡渣复合材料的冷结晶峰温时,样品的发泡倍率能够由7.24倍提高至9.33倍。这与大量诱导冷结晶形成结晶区域增多而提高发泡倍率相关[13]。

样品,发泡温度/℃:(a)PLA,120 (b)PLA/咖啡渣,120 (c)PLA/MDI,120 (d)PLA/咖啡渣/MDI,120 (e)PLA,130 (f)PLA/咖啡渣,130 (g)PLA/MDI,130 (h)PLA/咖啡渣/MDI,130图5 PLA/咖啡渣复合材料发泡样品的SEM照片Fig.5 SEM photos of the PLA/coffee ground composites foams
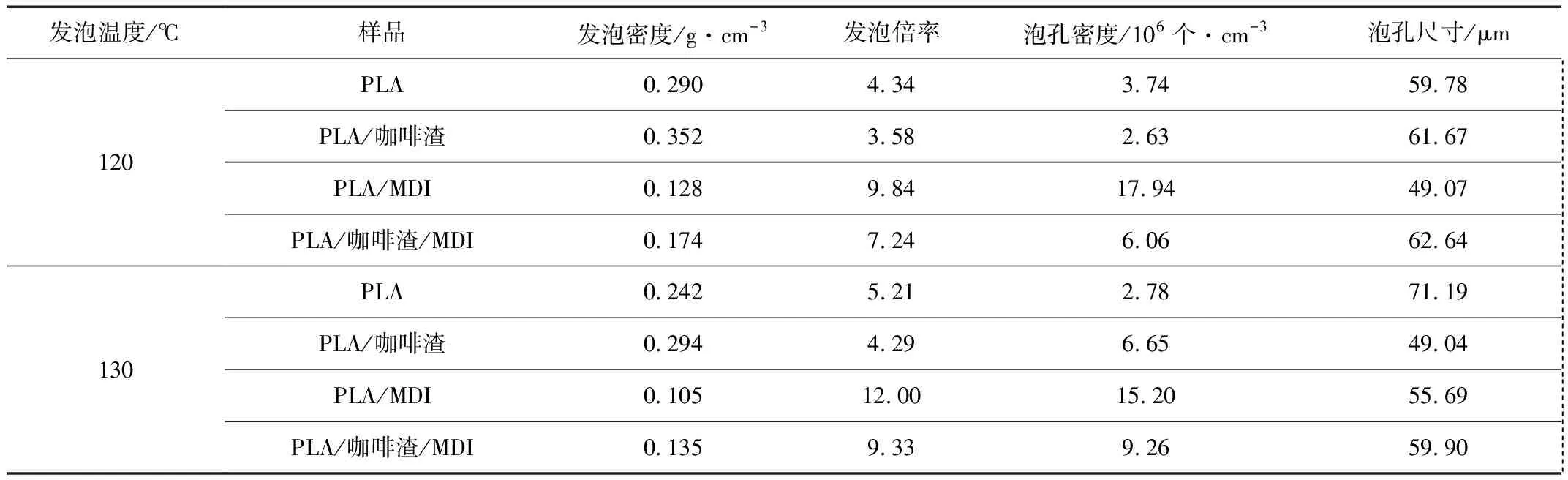
表3 PLA/咖啡渣复合材料发泡材料的泡孔密度和泡孔尺寸Tab.3 Cell density and cell size parameters of PLA/coffee ground composites foams
3 结论
(1)通过熔融共混法能够制备PLA/咖啡渣复合材料,加入MDI能够与PLA分子链反应起到扩链作用,有效提高复合材料的熔体弹性和末端松弛时间;
(2)加入MDI有助于PLA产生冷结晶和熔融双峰现象,同时提高其冷结晶度,诱导结晶作用显著影响了复合材料的力学性能;
(3)PLA/咖啡渣复合材料在诱导冷结晶的温度下发泡,其发泡倍率优于没有冷结晶的体系,MDI对PLA/咖啡渣复合材料的发泡过程和泡孔结构具有明显的调控作用。
参考文献:
[1] WANG X D, LIU W, ZHOU H F, et al. Study on the Effect of Dispersion Phase Morphology on Porous Structure of Poly (lactic acid)/Poly (ethylene terephthalate glycol-modified) Blending Foams[J]. Polymer, 2013, 54(21):5 839-5 851.
[2] ARRUDA L C, MAGATON M, BRETAS R E S, et al. Influence of Chain Extender on Mechanical, Thermal and Morphological Properties of Blown Films of PLA/PBAT Blends[J]. Polymer Testing, 2015(43):27-37.
[3] MUSSATTO S I, BALLESTEROS L F, MARTINS S, et al. Extraction of Antioxidant Phenolic Compounds from Spent Coffee Grounds[J]. Separation & Purification Technology, 2011, 83(1):173-179.
[4] SAFARIK I, HORSKA K, SVOBODOVA B, et al. Magnetically Modified Spent Coffee Grounds for Dyes Removal[J]. European Food Research & Technology, 2012, 234(2):345-350.
[5] LIU W, LI H Q, WANG X D, et al. Effect of Chain Extension on the Rheological Property and Thermal Beha-viour of Poly(lactic acid) Foams[J]. Cellular Polymers, 2013, 32(6):343-368.
[6] CHEN P, ZHOU H F, LIU W, et al. The Synergistic Effect of Zinc Oxide and Phenylphosphonic Acid Zinc Salt on the Crystallization Behavior of Poly (lactic acid)[J]. Polymer Degradation & Stability, 2015, 122:25-35.
[7] LIU W, WANG X, LI H Q, et al. Study on Rheological and Extrusion Foaming Behaviors of Chain-extended Poly (lactic acid)/Clay Nanocomposites[J]. Journal of Cellular Plastics, 2013, 49(6):535-554.
[8] HERRERA R, FRANCO L, PUIGGALI J. Characterization and Degradation Behavior of Poly(butylene adipate-co-terephthalate)s[J]. Journal of Polymer Science Part A: Polymer Chemistry, 2002, 40(23):4 141-4 157.
[9] NAJAFI N, HEUZEY M C, CARREAU P J. Polylactide (PLA)-clay Nanocomposites Prepared by Melt Compounding in the Presence of a Chain Extender[J].Composites Science & Technology, 2012(72): 608-615.
[10] SANTIS F D, PANTANI R, TITOMANLIO G. Nucleation and Crystallization Kinetics of Poly(lactic acid)[J]. Thermochimica Acta, 2011, 522(1/2):128-134.
[11] DING W D, KUBOKI T, WONG A, et al. Rheology, Thermal Properties, and Foaming Behavior of High D-content Polylactic Acid/Cellulose Nanofiber Composites[J]. Rsc Advances, 2015, 5(111):91 544-91 557.
[12] LIANG J Z. Reinforcement and Quantitative Description of Inorganic Particulate-filled Polymer Composites[J]. Composites Part B:Engineering, 2013, 51(4):224-232.
[13] LIU W, CHEN P, WANG X D, et al. Effects of Poly(butyleneadipate-co-terephthalate) as a Macromolecular Nucleating Agent on the Crystallization and Foaming Behavior of Biodegradable Poly(lactic acid)[J]. Cellular Polymers, 2017, 36(2):75-96.
猜你喜欢
包装工程(2022年1期)2022-01-26 09:03:10
科教导刊·电子版(2021年6期)2021-05-06 05:05:14
工程塑料应用(2020年11期)2020-11-28 01:57:50
中国塑料(2016年4期)2016-06-27 06:33:48
中国塑料(2016年3期)2016-06-15 20:30:01
华东理工大学学报(自然科学版)(2015年3期)2015-11-07 09:17:13
中国塑料(2015年7期)2015-10-14 01:02:40
合成材料老化与应用(2015年4期)2015-07-25 10:45:44
新疆钢铁(2015年3期)2015-02-20 14:13:56
中国塑料(2014年1期)2014-10-17 02:46:37
