
BIA 10- 2474药物临床研究中5例健康受试者神经系统损害及死亡事件的启示
2018-05-24 02:33陈霞
协和医学杂志 2018年3期
陈 霞
首都医科大学附属北京天坛医院国家神经系统疾病临床医学研究中心-临床试验中心, 北京 100050
2016年1月,正在法国雷恩进行的BIA 10- 2474 Ⅰ期临床试验传来噩耗,6名接受活性试验药给药的健康受试者中5人先后发生了不同程度的神经系统不良事件,最严重者脑死亡[1]! 消息传来,业界哗然,因为这是继2006年CD28激动剂TGN1412首次人体试验导致6名健康受试者严重致残事件后,又一起早期药物临床试验使健康受试者致死或致残的严重安全性事件。
健康受试者早期药物临床试验是否具有很高的风险?不同时期、不同地区的多个综述[2- 5]均表明,以健康受试者为对象的药物早期临床研究的不良事件发生率约为15%左右,其中活性药组的不良事件发生率约比安慰剂组高0.5~1倍。头痛是最常报告的不良事件,约占不良事件总数的2%。超过99%的不良事件为轻度或中度,严重不良事件非常少见,很少有致死或危及生命的情况。也许正因为此数据,每一个在健康受试者药物临床研究中发生的严重致残或致死性医疗事件,均会引起轰动效应。抱着“亡羊补牢”的心理,人们努力探寻这些事件的原因,总结教训,尽可能从系统上降低类似悲剧重现的风险。本文梳理BIA 10- 2474事件的发生过程,以期为控制早期药物临床试验风险带来些许启示。
1 事件回顾
1.1 BIA 10- 2474的作用机制
BIA 10- 2474是脂肪酸酰胺水解酶(fatty acid amide hydrolase,FAAH)抑制剂。FAAH是内源性大麻素的降解酶,因此BIA 10- 2474实际上是内源性大麻素系统的激活剂。BIA 10- 2474以共价结合的方式封闭FAAH的活性位点,其作用接近于不可逆抑制。因此,BIA 10- 2474的作用强度更依赖于峰浓度,抑制持续时间与FAAH本身的更新速率相关,与药物浓度达到最小抑制浓度的时间无关。
大麻素系统激活所产生的效应与吸食大麻类似,可引起欣快感、刺激食欲,还有止吐、镇痛、催眠等作用。大麻的合成类似物dronabinol(Marinol®)和nabilone (Cesamet®)已在一些国家上市,用于治疗癌症或艾滋病患者的呕吐和厌食症状。BIA 10- 2474是作为镇痛药被开发的,目前市场上尚无作用于大麻素系统的镇痛药。辉瑞和强生公司曾分别开发过以FAAH为靶标的镇痛药,这两种化合物对FAAH均具有很好的选择性,药效也是BIA 10- 2474的数百倍,且均完成了临床Ⅰ期研发,未发现严重毒性。遗憾的是,在Ⅱ期概念验证研究中,均因这两种药疗效欠佳而中止了研发。因此,BIA 10- 2474的作用机制还未在人类疼痛患者中被验证过。在这种情况下,再次基于同一机制、研发在靶标选择性和靶标作用强度上均未改进的新化合物,研发失败的概率无疑很高;且因为特异度欠佳,BIA 10- 2474还有可能在药理学有效剂量下引起抑制FAAH之外的脱靶(off-target)效应,带来潜在的安全性风险。
1.2 事件概况
此次发生死亡事件的研究是BIA 10- 2474的第3个人体研究。此前,研究者已先后完成了单次给药剂量递增研究(剂量0.25~100 mg,共入组64名受试者)和进食对药代动力学影响的研究(剂量40 mg,共12名受试者),前两项研究中并未出现严重或值得关注的不良事件。在进行多次给药剂量递增研究时,前4个剂量组亦未发生严重不良事件(图1)。
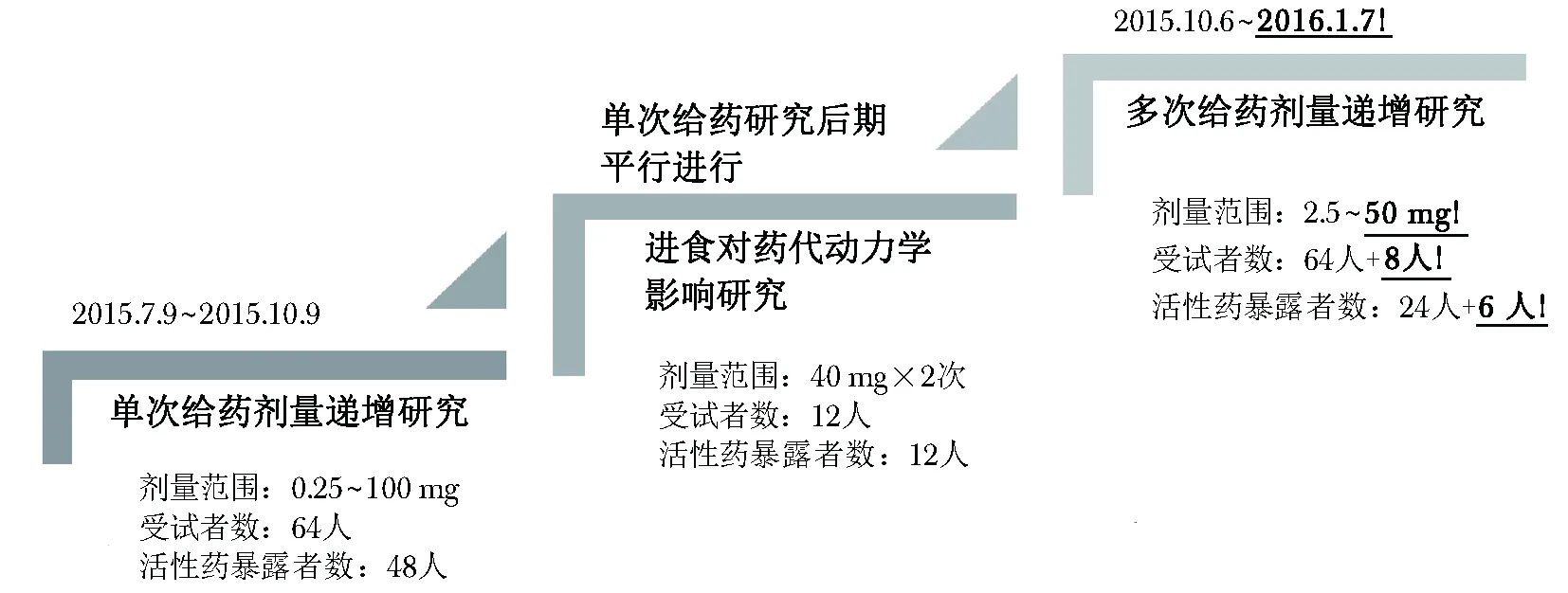
图1BIA 10- 2474 Ⅰ期临床试验流程
注:图中下划线加粗部分为发生神经系统不良事件和死亡事件的时间、剂量及对应组别的受试者人数
多次给药剂量递增研究于2016年1月6日爬坡至第5个剂量组(50 mg组),共纳入8名受试者,其中6人被随机分配接受BIA 10- 2474给药,2人接受安慰剂给药。根据方案,每名受试者在研究期间需服用10次研究药物,每天一次,每次服用50 mg BIA 10- 2474或外观相同的安慰剂。2016年1月10日(第5次服药后)的晚上,1名BIA 10- 2474组受试者(总服药剂量250 mg)因出现严重的神经系统症状(具体信息未披露)住院。次日,研究者仍继续安排剩下的7名受试者接受第6次给药(BIA 10- 2474组的总服药剂量达300 mg),更多BIA 10- 2474组受试者出现了类似神经系统症状,并相继住院。磁共振成像显示,5名(83.3%)BIA 10- 2474组受试者出现了不同程度的脑桥和海马病变。研究者遂中止了第7次给药。
2 流程和伦理风险管理与控制
BIA 10- 2474试验遵循新药早期研发的循序渐进原则,按药物暴露量和暴露时间递增顺序依次进行单次给药剂量递增试验、进食影响试验和多次给药剂量递增试验。在进行单次给药剂量递增试验时,研究者还采用了“哨兵法”给药设计,即每一剂量组分为两批给药,第1批各1名受试者接受BIA 10- 2474或安慰剂给药,待确认安全后,第2批剩下5名BIA 10- 2474组受试者和1名安慰剂组受试者再给药。鉴于单次给药剂量递增试验到最大爬坡剂量100 mg均未发生严重不良事件,多次给药剂量递增研究未再设计 “哨兵法”给药,而是在每一组给药结束后对受试者的药物暴露水平进行检测和推算,如果发现未来给药组的药物暴露水平可能远远超出前期研究中的暴露水平,则酌情修订方案规定的给药剂量。
在BIA 10- 2474事件中,研究者在首例严重不良事件发生后的处理显然不够谨慎。在1名受试者突然出现原因不明的急性病症并可能与研究药物相关的情况下,研究者未及时中止整个试验,对该例严重不良事件进行深入的病因分析和进展追踪,而在第2天继续给剩下7名受试者用药,做法欠妥,这使剩下的BIA 10- 2474组受试者继续暴露于未知风险中。另外,发生严重不良事件后,研究者应立即报告伦理委员会。目前披露的信息未体现首例事件发生后研究者与伦理委员会的互动,但2016年1月10日是周日,笔者推断即使研究者立即以传真形式报告了此例严重不良事件,伦理委员会也来不及在次日上午给药前进行审查和批复。按照目前的流程规定,研究者无需等到伦理委员会批复即可自行作出判断,决定是否继续研究。对于新药早期研究,是否应加强伦理对研究者的监管,强化突发严重不良事件后的紧急沟通,值得商榷。
3 研究设计风险管理与控制
3.1 给药剂量设置
创新药早期临床研究的目的之一是探索药物在人体应用的安全剂量范围,又称耐受剂量范围。一方面,研究者希望找到尽可能宽的耐受剂量范围,为后期药物研发和未来临床应用提供一定的探索/调整空间;另一方面,为了避免受试者遭受严重的致残或致死性伤害,研究者常常在研究中设置严格的安全性监控计划,根据安全性发现提前终止剂量爬坡的判定标准及风险预案,以便在药物风险初露端倪时及早中止研究给药或剂量递增,保护受试者安全。即便如此,仍然难以规避某些研究给药后快速出现的普遍性严重不良事件,故近年来早期临床试验中的剂量设置问题越来越受到重视,这类研究设计要求研究者综合考虑科学目的、受试者的安全性和试验的可实施性。
创新药早期临床研究,特别是首次人体研究剂量设置最常用的方法是借鉴前期的动物研究结果。2005年美国食品药品监督管理局(Food and Drug Administration,FDA)颁布了创新药首次人体研究的剂量设置指南。指南中,FDA建议将动物长期给药(通常为28 d或3个月连续给药)毒性研究中给药后未观察到显著毒性的剂量,即未观察到有害作用水平(no observed adverse effect level,NOAEL)用于人体研究中起始剂量的估算。为了避免种属间差异带来的误估风险,该指南还建议至少进行两种动物长期给药毒性研究,以获得不同动物的NOAEL剂量。按照FDA指南所提供的经验性公式,人们可计算动物NOAEL剂量所对应的人体等效剂量,从中选择最小剂量,再除以安全系数10,即可得到人类的最大推荐起始剂量。换句话说,创新药人类研究的起始剂量不能超过其在较敏感动物(即NOAEL剂量较低动物)中NOAEL人体等效剂量的1/10。
这种仅考虑毒性的估算方法很快就被发现存在漏洞。2006年,在CD28受体激动剂TGN1412的首次人体研究中,6名健康受试者在第一个剂量单次给药后纷纷出现严重不良事件并终身致残。有关专家立即对该事件展开了调查[6- 7],结果发现TGN1412的起始剂量设置完全符合FDA指南;出于谨慎,研究者甚至将安全系数扩大至160,即起始剂量仅相当于NOAEL人类等效剂量的1/160。但TGN1412的人类首次给药剂量仍远远高于该药的药理学起效剂量。在首次给药后,受试者体内的CD28被大量激活,形成免疫风暴并导致了严重不良反应。为此,欧洲药品管理局在TGN1412事件后颁布了新的指导原则[8],建议设置创新药首次人体研究的起始剂量时,除从安全性角度估算NOAEL人类等效剂量外,还需考虑药理学起效剂量,从低于或等于药理学起效剂量水平开始给药。以TGN1412为例,如果研究者当时考虑了药效因素,按最小预期生物学效应水平估算人类起始剂量[6,9],这场悲剧也许可以避免。另外,TGN1412事件还使人们加强了对全新机制药物的谨慎度,“哨兵法”给药即是在TGN1412事件后出现的一种控制受试者风险的设计方法。
3.2 BIA 10- 2474临床前研究的相关考虑
BIA 10- 2474在临床前仅做了两个药效学试验。这两个试验均未设计阳性对照,即研究者只能通过试验组模型动物的行为学反应及其与安慰剂组的差异,间接获得BIA 10- 2474具有镇痛效应的结论,此证据相对较弱。此外,研究还发现[1],人体完全抑制FAAH所需的BIA 10- 2474剂量约为1.25~5 mg,是致死事件发生时BIA 10- 2474给药剂量(50 mg)的1/10~1/40。虽然临床前研究的药效未必等同于患者真实情况,但过度向上递增、探索最大耐受剂量的必要性有待商榷。
BIA 10- 2474共对4种动物(大鼠、小鼠、犬和猴)进行了毒理学研究,这些毒性研究并未发现BIA 10- 2474的特异度毒性,对人类可能出现的毒性也无潜在提示[10],由动物研究所确定的NOAEL剂量均是合理的。
动物研究找到了BIA 10- 2474的4种代谢产物,暴露量均很低,约为母药的1%。鉴于代谢产物的暴露比例未达到相关指南规定的必须进行毒性研究的水平,临床前研究未针对这些代谢产物进行毒性研究[11]。
3.3 BIA 10- 2474人体研究的相关考虑
早期人体研究的目的之一是探索BIA 10- 2474的耐受剂量范围,未来临床研究将在此剂量范围内探索有效剂量。由于系统暴露是药物发挥药效和引起不良反应的物质基础,早期研究中还会考察药物的药代动力学特征,包括峰浓度、达峰时间、半衰期、剂量-暴露关系等。
BIA 10- 2474的单次最大给药剂量高达100 mg,受试者未出现严重不良反应。前期研究中服用BIA 10- 2474的76名受试者共报告了18个轻度不良事件,其中11个为心血管不良事件,其他为头晕、头痛、视物模糊或复视等。在发生死亡的多次给药剂量递增研究中,前4个剂量组的不良事件类型与前期试验相近。考虑到严重不良事件中的海马和脑桥病变在患病初期有可能症状隐匿,不易察觉,研究者事后联系了此前服用过BIA 10- 2474的所有受试者,安排其接受全面的神经系统临床评估和头颅磁共振成像检查,受试者中74%(62/84)完成了检查,但临床和影像学表现均未发现显著异常[1]。
单次给药剂量递增试验显示,BIA 10- 2474的暴露随剂量呈非线性递增,在40~100 mg之间出现清除饱和,即剂量递增100%,系统暴露的增幅超过100%,这意味着受试者在高剂量下系统暴露有可能超过动物毒性研究探测到的最大耐受暴露水平。连续给药的情况下,由于药物蓄积和时间依赖性代谢酶抑制等因素,这种非线性暴露递增可能更加显著,进一步增加受试者暴露于毒性浓度的风险。人类单次给药剂量递增研究中还发现了4种含量很低的代谢产物,尽管其暴露水平未达到指南规定的代谢产物研究标准(10%),但已知其中有一种代谢产物的毒性非常高,即使浓度很低也可引起严重的毒性反应。
以上情况提示,BIA 10- 2474多次给药试验在50 mg剂量组出现的严重神经系统不良事件具有突发性。既不曾在以往服药的受试者中观察到,也未在受试者前期服药过程中显露征兆。BIA 10- 2474在50 mg剂量附近出现的非线性暴露递增可能对此有一定贡献,而前期的“无迹可寻”也在一定程度上影响了研究者对首例受试者住院病因的判定和处理。
4 启示
本文根据BIA 10- 2474专家调查组的报告[1]整体梳理了这个悲剧性事件的前因后果。虽然目前还未完全找到BIA 10- 2474事件的直接原因,但现有信息已给我们带来了深刻启示与反思。
第一,应在药物进入临床研发前根据已获得的研究数据和同类药物的相关信息审慎决策,以便提高研发成功率,并为临床试验受试者规避不必要的风险。
第二,在设计方面,不仅应考虑药物的临床前安全性特征,还应考虑药物的药效特征,在兼顾受试者安全的前提下达成试验的科学目的。
第三,在进行高风险新药早期临床研究期间,应始终保持高度警惕性,不放过任何可能提示药物风险的信号,遇到问题全面分析,保守处理。
吃一堑,长一智。研究者不会因为某个悲剧事件停止创新药研发的脚步,但应由此认识到自身知识和经验的局限性,注重与不同领域专家密切合作,理性决策,谨慎实施!
参 考 文 献
[1] Kaur R, Sidhu P, Singh S. What failed BIA 10- 2474 Phase I clinical trial? Global speculations and recommendations for future Phase I trials[J]. J Pharmacol Pharmacother, 2016,7:120- 126.
[2] Zarafonetis CJ, Riley PA Jr, Willis PW 3rd,et al. Clinically significant adverse effects in a Phase 1 testing program[J]. Clin Pharmacol Ther, 1978,24:127- 132.
[3] Sibille M, Deigat N, Olagnier V, et al. Adverse events in phase one studies: a study in 430 healthy volunteers[J]. Eur J Clin Pharmacol, 1992,42:389- 393.
[4] Lutfullin A, Kuhlmann J, Wensing G. Adverse events in volunteers participating in phase I clinical trials: a single-center five-year survey in 1,559 subjects[J]. Int J Clin Pharmacol Ther,2005,43:217- 226.
[5] Johnson RA, Rid A, Emanuel E, et al. Risks of phase I research with healthy participants: A systematic review[J]. Clin Trials, 2016,13:149- 160.
[6] Kenter MJ, Cohen AF. Establishing risk of human experimentation with drugs: lessons from TGN1412[J]. Lancet,2006, 368:1387- 1391.
[7] Guideline on strategy to identify and mitigate risks for first-in-human clinical trials with investigational medical products[EB/OL]. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/07/WC500232186.pdf.
[8] Tranter E, Peters G, Boyce M, et al. Giving monoclonal antibodies to healthy volunteers in phase 1 trials: is it safe? [J]. Br J Clin Pharmacol, 2013,76:164- 172.
[9] Hackam DG, Redelmeier DA. Translation of research evid-ence from animals to humans[J]. JAMA, 2006,296:1731- 1732.
[10] Whitebread S, Dumotier B, Armstrong D, et al.Secondary pharmacology: screening and interpretation of off-target activities-focus on translation[J]. Drug Discov Today,2016,21:1232- 1242.
[11] Ahuja V, Sharma S.Drug safety testing paradigm, current progress and future challenges: an overview[J]. J Appl Toxicol, 2014,34:576- 594.
猜你喜欢
中国心血管杂志(2022年2期)2022-11-25
中国心血管杂志(2022年4期)2022-11-25
当代水产(2021年6期)2021-08-13
河池学院学报(2021年1期)2021-07-10
中国心血管杂志(2021年6期)2021-01-02
小哥白尼(野生动物)(2019年5期)2019-08-27
英语文摘(2019年2期)2019-03-30
中国心血管杂志(2019年3期)2019-01-04
中成药(2018年11期)2018-11-24
中华手工(2018年6期)2018-07-17
