
基于诱导多能干细胞技术的罕见病细胞模型及其应用
2018-05-24 05:10:58崔亚洲韩金祥
协和医学杂志 2018年3期
时 良,崔亚洲,韩金祥
1山东省医学科学院 山东省医药生物技术研究中心,济南 250062 2山东省罕见疾病防治协会,济南 250062
罕见病通常是指患病率低但却严重威胁生命的疾病。罕见病的定义在立法和政策上有所不同,通常以疾病发生率或患病率作为标准或临界值。根据流行病学和基因组学数据,美国国立卫生研究院估计世界各地大约有7000种罕见病。中国常用世界卫生组织对于罕见病的定义,即患病率低于0.65‰~1‰,然而,这些定义有一个相对宽泛的范围。目前,对于中国罕见病的定义尚未统一[1]。尽管如此,中国作为人口大国,罕见病患者人群庞大,很多罕见病发病原因未知,且无有效根治手段,因此基于分子机制的研究和开发具有重要意义。
超过80%的罕见病与遗传密切相关[2]。诱导多能干细胞(induced pluripotent stem cells,iPSCs)技术可以产生疾病特异性iPSCs,进而分化为与疾病相关的功能细胞用于构建疾病模型,体外再现疾病表型、模拟遗传学变化和病理过程,在此基础上研究发病机制、筛选安全有效的药物、替换患病的细胞或组织等。最重要的是,iPSCs技术不像胚胎干细胞(embryonic stem cells,ESCs)研究一样饱受伦理学争议和免疫排斥的困扰,广大研究者基于此项技术已成功建立多种罕见病模型,并进行了深入研究。本文主要讨论了iPSCs技术应用于罕见病疾病模型的建立及其在药物筛选、细胞治疗方面的应用。
1 利用诱导多能干细胞技术建立罕见病疾病模型的优势
传统罕见病病因和病理生理机制研究往往依赖于原代或患者来源的永生化细胞系。虽然原始细胞类型很容易从血液或组织活检中获得,但与疾病相关的细胞类型如涉及大脑或心脏的细胞不易获得,也不可能无限期增殖。此外,永生化细胞系随着培养时间的延长往往不能准确反映原代细胞的状态,限制了其在功能研究中的可靠性。同样,动物模型虽然是体内研究不可替代的工具,但动物和人类之间有相当多的解剖学、胚胎学和代谢差异,如最常用的动物模型小鼠与人类在心脏大小和静息心率等方面均不同[3];又如将转基因小鼠模型应用于阿尔茨海默病的研究中,由于人类和小鼠神经细胞间的种属差异造成大量神经元缺失,因而无法准确再现人的病理学过程[4],成为基础研究转化为临床试验的最大障碍。因此,迫切需要建立人类疾病模型来弥补生物医学研究中使用动物模型的缺陷。
人iPSCs技术的诞生为建立疾病模型提供了更多选择。人iPSCs与ESCs高度相似,如可显示ESCs样的细胞形态、表达多能干细胞标志物、拥有相似的基因表达和表观遗传学状态且具有体内外分化成三胚层的能力[5- 6]。多种易获得的供体细胞可重编程为人iPSCs(表1),其可分化为各种功能细胞,如心肌细胞、肝细胞等较难从患者身上直接获取的疾病相关细胞。研究发现,iPSCs中残存对供体细胞的表观记忆,影响了iPSCs下游分化[7- 8]。但在众多研究中,同时用人ESCs与iPSCs建立疾病模型可在体外得到相似的表型,证明了iPSCs技术用于建立疾病模型的可行性[9]。为了iPSCs的后期应用,重编程方法不断改进,可采用安全高效、非病毒整合的方法(表2)。
在既往罕见病研究中,将患者特异性的iPSCs或功能细胞与健康人进行比较,然而由于细胞来自不同个体,拥有不同的遗传背景和基因表达水平,在一定程度上会干扰实验结果,可能会影响后期临床应用。随着基因编辑技术的快速发展,借助于基因打靶技术修正缺陷的基因产生遗传学匹配的对照细胞用于建立疾病模型,避免细胞系不同带来的遗传背景差异或偶然性结果[36- 37]。由于罕见病患者分布分散,不易获取供体细胞,iPSCs结合基因编辑技术亦可为研究不易获得的罕见病或基因型开辟新的途径。利用CRISPR/Cas9基因编辑工具能有效引入特定的突变位点,仅影响某个基因的一个副本,即患者的iPSCs仅在特定位点与正常对照组iPSCs不同,由此可获得携带致病基因的iPSCs[38- 39]。

表 1 产生人诱导多能干细胞的多种供体来源
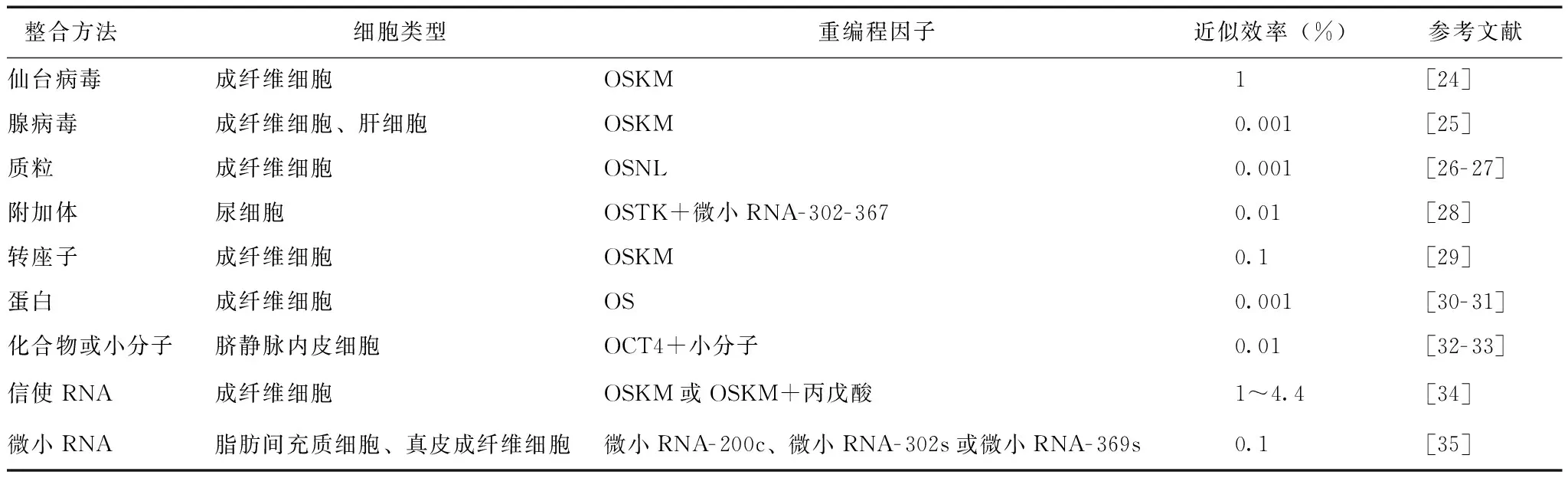
表 2 采用非病毒整合方法获得诱导多能干细胞
K:KLF4;L:LIN28;M:c-MYC;O:OCT4;S:SOX2
iPSCs技术结合基因编辑技术对于研究表型差异较小的散发性或自发性疾病尤为重要[40]。常用两种分析方法来避免除目的基因外的基因多样性干扰,在疾病特异性iPSCs中修正疾病相关基因或在野生型iPSCs中引入致病突变[41]。研究人员利用这种方式建立了C1型尼曼匹克症疾病模型,与对照组相比,疾病特异性iPSCs分化而来的肝细胞和神经细胞表现出疾病相关缺陷,比如胆固醇积累和异常的细胞自噬现象,通过修正突变基因NPC1,修复了上述缺陷[42],因此iPSCs技术为罕见病研究开辟了新的途径。随着iPSCs技术的不断发展,患者特异性iPSCs的获取将更加规范化和规模化,疾病模型会成为一种有效的工具,为新药筛选、个性化再生医疗方案的制定另辟蹊径。
2 基于诱导多能干细胞罕见病疾病模型的建立及机制研究
细胞重编程可产生基于iPSCs的罕见病疾病模型,从根本上推动了对罕见病病理生理学和发病机制的深入理解,尤其是先前不易获得的细胞或组织如心肌细胞、神经细胞等;另外,iPSCs结合基因编辑技术,可解决罕见病患者样本难获得问题。迄今为止,绝大多数疾病模型针对以孟德尔遗传方式导致的病例而建立,但仍有很多疾病是偶发性或由多个位点基因的多态性导致。转基因动物或细胞系等传统建模方法极具挑战性,而iPSCs技术可解决这一难题,即便有多个未知突变,疾病特异性iPSCs携带患者遗传背景仍可再现病理表型,并在此基础上探究疾病发生的机制。最近研究者利用iPSCs技术成功建立了多种罕见病疾病模型,并发现了其重要机制或靶点(表3)。
2.1 单基因罕见病
单基因突变造成的疾病称为单基因病,单基因缺陷引起的新生儿患病率通常小于0.1‰,因而绝大多数属于罕见病。iPSCs技术被广泛应用于建立先天突变的单基因罕见病模型,研究发现多种单基因疾病来源的iPSCs通过合适的下游分化方法得到疾病相关细胞类型,可在体外真实地模拟疾病病理学过程[62- 63]。通过iPSCs技术可建立单基因罕见病疾病模型,例如与神经损伤有关的疾病(弗里德赖希共济失调、共济失调性毛细血管扩张症、C1型尼曼匹克症、柯凯因综合征等),采用适宜的下游分化方法可得到疾病相关功能细胞,深入探究疾病发病机制[64- 67]。随着iPSCs技术不断发展,关于神经发育障碍罕见病,最近研究者发现了一种更加高效的建模方法,从尿液收集到获得神经亚型仅需75 d[68]。
脊髓性肌萎缩症是早发性罕见病,由于运动神经元1突变导致,将患者特异性iPSCs分化为神经细胞用于模型建立[62]。运动神经元1突变导致运动神经元衰退和肌肉萎缩变性。Ⅰ型脊髓性肌萎缩症患者通常在出生6个月内表现出症状,随着病情加重, 2岁时患儿死亡[69]。最初的iPSCs疾病模型,将Ⅰ型脊髓性肌萎缩症患者成纤维细胞来源的iPSCs分化成运动神经元[62]。从患者特异性iPSCs分化而来的运动神经元与正常组相比存活能力降低。这项研究结果表明,患者来源的iPSCs可用于早发性单基因罕见病模型建立。
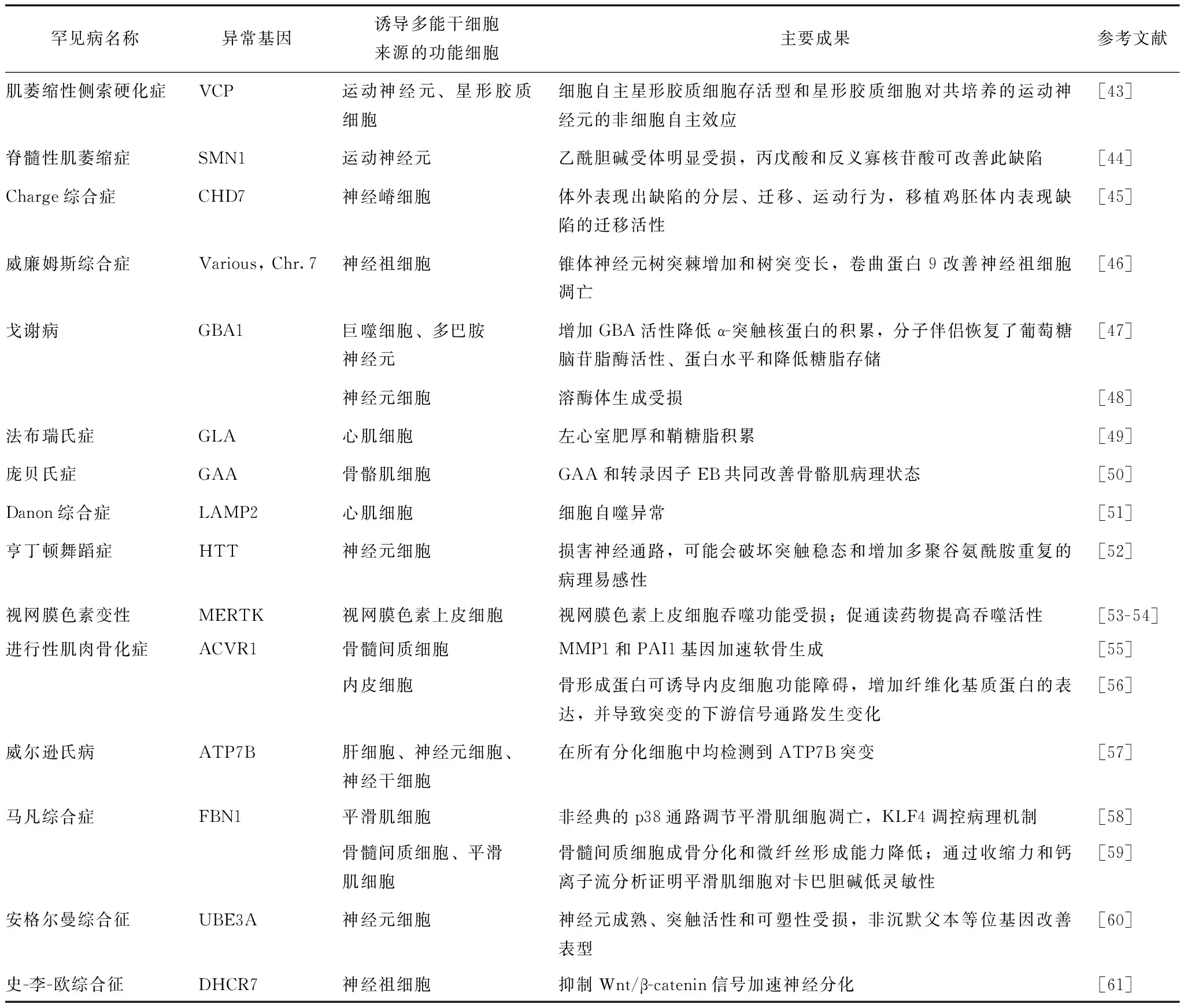
表 3 利用诱导多能干细胞技术建立的罕见病疾病模型
2.2 多基因罕见病
在染色体区域的缺失或重复称为多基因病,复杂罕见病通常涉及多个或未知的异常基因。如自闭症因表型和病因异质性,建立细胞模型或动物模型存在很大困难,因而揭示潜在的遗传学、病理生理学机制具有很大挑战。iPSCs技术的出现对于理解复杂或散发性罕见病带来了新的希望[70],研究人员假设自闭症患者中增多的脑体积和神经元数量可能是神经祖细胞增殖率增加的结果[71],于是将患者特异性iPSCs分化为神经祖细胞,患者的神经祖细胞比正常组增殖速度快,与假设一致。神经元则表现出异常神经发生和突触减少,导致神经元网络的功能缺陷。在哺乳动物大脑发育过程中,神经祖细胞是受到严格调控的,如果异常增殖会导致长久性分化异常和缺陷,将导致自闭症的发生。第一型类胰岛素生长因子(insulin-like growth factor 1,IGF- 1)是一种自然产生的神经营养因子,对大脑发育和可塑性至关重要[72- 73]。研究人员发现IGF- 1可改善神经网络异常,目前正处于临床试验阶段。
2.3 迟发性罕见病
相比于早发疾病,细胞的老化程度是迟发疾病病理学研究的重要因素,建立迟发性罕见病模型更具挑战性,因为iPSCs的特点是具有早期胚胎的基因表达程序[74]。成功建模的重要方法是人工诱导细胞的衰老[75- 76]。具体方法如下:(1)氧化应激:使用MG- 132和吡唑醚菌酯等化合物,通过靶向线粒体的功能或蛋白质降解途径,进而促进细胞衰老[77- 78];(2)早衰蛋白:由核纤层蛋白截短后产生,可导致过早老化[75]。
最近发现的一种更具生理学意义的方法是采用小分子抑制端粒酶活性,其显示了衰老的经典特征,包括DNA损伤、活性氧增加,酪氨酸羟化酶的下调[79]。早衰综合症的iPSCs模型,如哈钦森-吉尔福德早衰综合症,不仅成功地模拟了高速分化和衰老的干细胞,而且也促进了与年龄有关的标志物应用于更普遍的迟发性疾病如帕金森病研究[80]。
迄今为止,多种疾病的建模方案是基于单一的功能细胞。然而,对于很多疾病而言,不止一种功能细胞能准确模拟疾病发生,如自闭症建模,使用的是患者特异性神经祖细胞和神经元细胞。除此之外,为了更精确地再现疾病表型,需将不同类型的细胞共培养以研究细胞间的相互作用,如3D组织培养技术。从传统的二维分化培养方法逐步发展为空间模拟人类组织或器官的交互作用。目前采用老鼠和人的组织干细胞或多能干细胞可产生多种器官,包括大脑、视网膜、肠道、肾脏、肝脏、肺、胃[81]。人iPSCs分化形成的组织因与内源性细胞组织或器官结构相似,而被广泛应用于模拟人类生理和发育过程中细胞间相互作用。以组织形式存在的多种细胞比单独分化得来的细胞功能上更加成熟,主要原因是存在细胞间的通讯,比如3D结构中神经元细胞和星形胶质细胞。目前已被用于模拟人类器官形成和疾病病理过程,检验可用于治疗的化合物或进行细胞移植[82- 84]。3D组织培养技术有待发掘更加标准化的培养条件和胞外基质,高效地再现组织系统,将更适用于建立精准疾病模型,进而筛选新药,促进再生医学发展[85]。
3 基于诱导多能干细胞罕见病模型的应用
iPSCs技术除了上述提到的建立罕见病模型,在发病机制和功能上进行深入探究外,在药物筛选、临床治疗等方面亦具有广泛应用。在有效疾病模型的基础上,探究罕见病的病理生理学进程,利用功能细胞筛选新型药物,可推进细胞或组织治疗等再生医学的发展,达到干细胞研究的最终目的。结合基因编辑技术,具体应用流程如图1所示,重编程罕见病患者的体细胞,诱导得到患者特异性iPSCs,结合基因编辑工具修正患病基因得到同型对照组,两组细胞系均分化产生大量的功能细胞,在体外再现疾病表型,在此基础上发现新的诊断标志物、筛选安全有效的药物、替换患病的细胞或组织。这种定制化的治疗方法可以避免免疫排斥和伦理学争议。
3.1 药物筛选
罕见病的药物开发过程与普通疾病相似,需要大量资源,通常持续10~12年。952种中国住院患者可见的罕见病中,50种罕见病有相应的孤儿药在中国上市、获得临床批件或正在进行临床试验,95%的罕见病目前尚无特效治疗药物,患者预后不佳,给社会带来沉重负担。因罕见病患者基数小而限制了全面的药物试验,因而准确测定患者药物反应和新陈代谢情况以降低不良反应对罕见病患者的治疗至关重要。考虑到整体市场份额不足,药品开发对制药公司通常缺乏吸引力。直到1983年,《孤儿药法案》才将美国食品药品监督管理局(Food and Drug Administration,FDA)批准的罕见病药物份额提高至35%左右[2]。基于iPSCs的罕见病模型为发现罕见病可用药物带来了新的希望。家族性自主神经功能障碍是单基因遗传的早发性疾病,影响神经嵴细胞系,由IKBKAP编码核因子抑制剂κB激酶复合物相关蛋白突变引起,表现为神经系统缺陷和小纤维感觉神经元功能障碍。在iPSCs模型的基础上,分选并纯化自主神经元的神经嵴细胞前体。研究小组检测了6912个小分子化合物,发现8个化合物可恢复IKBKAP的表达,其中SKF- 86466可改善异常剪接[86]。这是使用基于人iPSCs疾病模型进行高通量药物筛选的第一个研究成果。
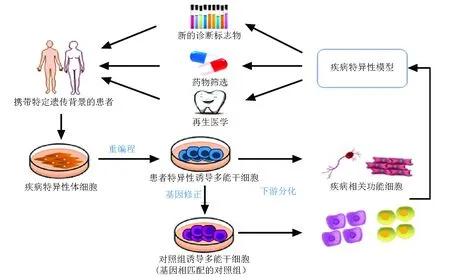
图1诱导多能干细胞技术用于罕见病研究流程图
疾病特异性iPSCs的另一个应用是药物重新定位,即在现有的已被批准用于特定疾病的药物中,找到其在其他疾病中新的应用。研究α- 1抗胰蛋白酶缺乏症的学者,从iPSCs分化得到肝细胞,利用已建立的临床化合物数据库,筛选了3131种临床批准的化合物(2800种药物已被美国FDA/国外同行批准或已进入第二阶段临床试验),确定5种可改善该疾病表型的临床药物,绕过临床前和临床研究早期阶段,直接作为临床疗法迅速进行临床试验[87]。软骨发育不全由成纤维细胞生长因子受体3突变导致,日本京都大学研究人员成功获得软骨发育不全患者的iPSCs,并在体外再现了异常的软骨形成过程;在此基础上,通过大量化合物筛选,研究者惊喜地发现他汀类药物可明显恢复骨骼生长,改善软骨发育不全的症状[88]。他汀类药物因有降低胆固醇的功效而被广泛用于治疗心血管疾病,如果临床试验成功,将意味着扩大了他汀类药物的适用范围。已批准的药物被确定可治疗其他疾病并可在临床试验中进一步评估,而无需长时间的临床前开发。此外,由于这些药物的作用靶点已知(如激酶抑制剂或蛋白酶),有助于其在新的疾病中确定新的药物靶点和治疗方法。
通过以上研究,证明了与使用永生化细胞系和动物模型相比,患者特异性iPSCs分化得到的疾病相关细胞更能准确反映药物的疗效。此外,已批准的药物通过iPSCs罕见病模型可发现新的适应症。
3.2 药物毒性试验
新药开发需要高额的经费,主要用于临床试验后期出现的无法预测的副作用[89]。然而,iPSCs模型能有效预测候选药物可能引起严重副作用,从而使候选药物在后期毒性试验中失败率大大降低。例如,西沙必利原本用于治疗胃食管反流病,因对心脏有毒性而退出市场。研究人员将遗传性长QT综合征、家族性肥厚性心肌病、家族性扩张型心肌病患者的iPSCs分化为心肌细胞,这些心肌细胞再现了疾病表型如电生理功能障碍,在此基础上用已知化合物包括西沙必利检测对心脏毒性的反应,发现患者来源的心肌细胞比正常组对西沙必利导致的异常更加敏感[90]。鉴于多种药物因对人体产生副作用而被撤出市场,可能是当前不全面的评估方法导致,iPSCs模型可进一步证实和补充。
3.3 细胞治疗
利用iPSCs技术促进再生医学(内源再生过程或细胞移植后替代受损组织)的发展引起了人们极大兴趣。2014年Takahashi研究小组进行了第一次iPSCs临床试验,其将老年性黄斑变性患者自身iPSCs来源的视网膜色素上皮细胞进行自体移植治疗,治疗结果为阳性,阻止黄斑变性并改善了患者的视力[91],虽然由于第2个患者iPSCs与供体成纤维细胞相比,存在3个单核苷酸变异和3个拷贝数变异而终止了下一步移植,但预计会重新开始细胞治疗[92]。基因、表观遗传、染色体的变化往往是由iPSCs体外培养导致[93],目前尚不完全清楚这些突变是否由重编程过程引起。尽管目前干细胞移植存在局限性,但其为罕见病治疗开辟了新的道路,而人类干细胞治疗罕见病的临床试验已经开始[94]。
利用iPSCs技术进行细胞治疗具有很多优势:(1)iPSCs能够自我更新,可获得足够数量的细胞,随后在体外可分化成任何类型的细胞;(2)iPSCs来源于患者自身供体细胞,避免寻找与组织相容性抗原兼容的细胞供体和使用免疫抑制剂。
然而,将iPSCs技术真正用于人类细胞治疗,仍有很多困难需要克服:(1)iPSCs的致瘤性[95],为确保最终分化得到的细胞中不含未分化的iPSCs,研究人员发现小分子抑制剂可进行筛选,使未分化的iPSCs死亡而不影响分化的细胞,降低了致瘤风险[96]。(2)由于iPSCs在体外培养时间较长,会导致异常核型和拷贝数变异[97],因此,在投入临床治疗前,需仔细排查iPSCs来源的细胞是否存在潜在的遗传变异风险,进行严格的鉴定以确保其纯度、质量和无菌。人类iPSCs平台与基因编辑、3D组织培养技术相结合,可让iPSCs技术为以干细胞为基础的细胞疗法提供更强大的细胞资源。
4 总结与展望
罕见病严重影响人类健康,相关研究已引起全球重视。尽管如此,罕见病仍然面临着难诊断、难治疗的挑战,并发症的存在亦增加了其诊治难度。为了推动中国罕见病政策的制定、深入发现遗传机制、提升临床诊疗水平,2016年“精准医学研究”重点专项“罕见病临床队列研究”的重要内容即是建立全国统一的罕见病注册登记系统,完善可共享的临床队列和样本库,整合临床诊疗信息,进而建立可开展预后研究的随访数据库体系。基于此,2017年3月1日山东省罕见病注册登记系统正式上线,目前已注册罕见病病例850例。在十三五国家重点研发计划“罕见病临床队列研究项目”的指导下,建立的中国国家罕见病注册体系,将为中国罕见病精准诊断与转化医学提供强大动力。
在此平台的支撑下,采用人的疾病模型来模拟这些复杂、多样的症状,可更深入剖析发病机制、开发新的治疗方法。iPSCs技术在罕见病建模方面具有独特优势,并成功建立了单基因、多基因、迟发性罕见病疾病模型,该模型已成为药物筛选、药物毒性研究、个性化细胞治疗的有效工具,推动了精准医学的发展。随着高通量测序、基因编辑技术和小分子筛选技术的突破,与这些技术结合起来,开发罕见病的治疗干预措施,将为罕见病患者的诊治带来前所未有的希望。
参 考 文 献
[1] Cui Y, Han J. Defining rare diseases in China[J]. Intractable Rare Dis Res, 2017, 6:148- 149.
[2] Melnikova I. Rare diseases and orphan drugs[J]. Nat Rev Drug Discov, 2012, 11:267- 268.
[3] Hamlin RL, Altschuld RA. Extrapolation from mouse to man[J]. Circ Cardiovasc Imaging, 2011, 4:2- 4.
[4] Onos KD, Sukoff Rizzo SJ, Howell GR, et al. Toward more predictive genetic mouse models of Alzheimer’s disease[J]. Brain Res Bull, 2016, 122:1- 11.
[5] Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors[J]. Cell, 2007, 131:861- 872.
[6] Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluri-potent stem cell lines derived from human somatic cells[J]. Science, 2007, 318:1917- 1920.
[7] Kim K, Zhao R, Doi A, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells[J]. Nat Biotechnol, 2011, 29:1117- 1119.
[8] Ohi Y, Qin H, Hong C, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells[J]. Nat Cell Biol, 2011,13:541- 549.
[9] Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery[J]. Nat Rev Mol Cell Biol, 2016, 17:170- 182.
[10] Wada N, Wang B, Lin NH, et al. Induced pluripotent stem cell lines derived from human gingival fibroblasts and periodontal ligament fibroblasts[J]. J Periodontal Res, 2011, 46:438- 447.
[11] Aasen T, Raya A, Barrero MJ, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes[J]. Nat Biotechnol, 2008, 26:1276- 1284.
[12] Giorgetti A, Montserrat N, Aasen T, et al. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2[J]. Cell Stem Cell, 2009, 5:353- 357.
[13] Haase A, Olmer R, Schwanke K, et al. Generation of induced pluripotent stem cells from human cord blood[J]. Cell Stem Cell, 2009, 5:434- 441.
[14] Seki T, Yuasa S, Fukuda K. Generation of induced pluripotent stem cells from a small amount of human peri-pheral blood using a combination of activated T cells and Sendai virus[J]. Nat Protoc, 2012, 7:718- 728.
[15] Simara P, Tesarova L, Rehakova D, et al. Reprogramming of adult peripheral blood cells into human induced pluripotent stem cells as a safe and accessible source of endothelial cells[J]. Stem Cells Dev, 2018, 27:10- 22.
[16] Aoki T, Ohnishi H, Oda Y, et al. Generation of induced pluripotent stem cells from human adipose-derived stem cells without c-MYC[J]. Tissue Eng Part A, 2010, 16:2197- 2206.
[17] Sugii S, Kida Y, Kawamura T, et al. Human and mouse adipose-derived cells support feeder-independent induction of pluripotent stem cells[J]. Proc Natl Acad Sci USA, 2010, 107:3558- 3563.
[18] Kim JB, Greber B, Arauzo-Bravo MJ, et al. Direct reprogramming of human neural stem cells by OCT4[J]. Nature, 2009, 461:649- 653.
[19] Cai J, Li W, Su H, et al. Generation of human induced pluripotent stem cells from umbilical cord matrix and amniotic membrane mesenchymal cells[J]. J Biol Chem, 2010, 285:11227- 11234.
[20] Li C, Zhou J, Shi G, et al. Pluripotency can be rapidly and efficiently induced in human amniotic fluid-derived cells[J]. Hum Mol Genet, 2009, 18:4340- 4349.
[21] Liu H, Ye Z, Kim Y, et al. Generation of endoderm-derived human induced pluripotent stem cells from primary hepato-cytes[J]. Hepatology, 2010, 51:1810- 1819.
[22] Zhou T, Benda C, Dunzinger S, et al. Generation of human induced pluripotent stem cells from urine samples[J]. Nat Protoc, 2012, 7:2080- 2089.
[23] Uhm KO, Jo EH, Go GY, et al. Generation of human induced pluripotent stem cells from urinary cells of a healthy donor using a non-integration system[J]. Stem Cell Res, 2017, 21:44- 46.
[24] Fusaki N, Ban H, Nishiyama A, et al. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome[J]. Proc Jpn Acad Ser B Phys Biol Sci, 2009, 85:348- 362.
[25] Stadtfeld M, Nagaya M, Utikal J, et al. Induced pluripotent stem cells generated without viral integration[J]. Science, 2008, 322:945- 949.
[26] Si-Tayeb K, Noto FK, Sepac A, et al. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors[J]. BMC Dev Biol, 2010, 10:81.
[27] Lorenzo IM, Fleischer A, Bachiller D. Generation of mouse and human induced pluripotent stem cells (iPSC) from primary somatic cells[J]. Stem Cell Rev, 2013, 9:435- 450.
[28] Xue Y, Cai X, Wang L, et al. Generating a non-integrating human induced pluripotent stem cell bank from urine-derived cells[J]. PLoS One, 2013, 8:e70573.
[29] Woltjen K, Michael IP, Mohseni P, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells[J]. Nature, 2009, 458:766- 770.
[30] Zhou H, Wu S, Joo JY, et al. Generation of induced pluripotent stem cells using recombinant proteins[J]. Cell Stem Cell, 2009, 4:381- 384.
[31] Kim D, Kim CH, Moon JI, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins[J]. Cell Stem Cell, 2009, 4:472- 476.
[32] Ichida JK, Blanchard J, Lam K, et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog[J]. Cell Stem Cell, 2009, 5:491- 503.
[33] Zhu S, Li W, Zhou H, et al. Reprogramming of human primary somatic cells by OCT4 and chemical compounds[J]. Cell Stem Cell, 2010, 7:651- 655.
[34] Warren L, Manos PD, Ahfeldt T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA[J]. Cell Stem Cell, 2010, 7:618- 630.
[35] Miyoshi N, Ishii H, Nagano H, et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs[J]. Cell Stem Cell, 2011, 8:633- 638.
[36] Kwart D, Paquet D, Teo S, et al. Precise and efficient scarless genome editing in stem cells using CORRECT[J]. Nat Protoc, 2017, 12:329- 354.
[37] Park CY, Sung JJ, Choi SH, et al. Modeling and correction of structural variations in patient-derived iPSCs using CRISPR/Cas9[J]. Nat Protoc, 2016, 11:2154- 2169.
[38] Paquet D, Kwart D, Chen A, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9[J]. Nature, 2016, 533:125- 129.
[39] Zhang Y, Schmid B, Nielsen TT, et al. Generation of a human induced pluripotent stem cell line via CRISPR-Cas9 mediated integration of a site-specific heterozygous mutation in CHMP2B[J]. Stem Cell Res, 2016,17:148- 150.
[40] Hockemeyer D, Jaenisch R. Induced pluripotent stem cells meet genome editing[J]. Cell Stem Cell, 2016, 18:573- 586.
[41] Suh W. A new era of disease modeling and drug discovery using induced pluripotent stem cells[J]. Arch Pharm Res, 2017, 40:1- 12.
[42] Maetzel D, Sarkar S, Wang H, et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells[J]. Stem Cell Reports, 2014, 2:866- 880.
[43] Hall CE, Yao Z, Choi M, et al. Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of VCP-related ALS[J]. Cell Rep, 2017, 19:1739- 1749.
[44] Yoshida M, Kitaoka S, Egawa N, et al. Modeling the early phenotype at the neuromuscular junction of spinal muscular atrophy using patient-derived iPSCs[J]. Stem Cell Reports, 2015, 4:561- 568.
[45] Okuno H, Renault Mihara F, Ohta S, et al. CHARGE syndrome modeling using patient-iPSCs reveals defective migra-tion of neural crest cells harboring CHD7 mutations[J]. Elife, 2017, 6.
[46] Chailangkarn T, Muotri AR. Modeling Williams syndrome with induced pluripotent stem cells[J]. Neurogenesis, 2017, 4:e1283187.
[47] Aflaki E, Borger DK, Moaven N, et al. A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and Parkinsonism[J]. J Neurosci, 2016, 36:7441- 7452.
[48] Awad O, Sarkar C, Panicker LM, et al. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells[J]. Hum Mol Genet, 2015, 24:5775- 5788.
[49] Chou SJ, Yu WC, Chang YL, et al. Energy utilization of induced pluripotent stem cell-derived cardiomyocyte in Fabry disease[J]. Int J Cardiol, 2017, 232:255- 263.
[50] Sato Y, Kobayashi H, Higuchi T, et al. TFEB overexpression promotes glycogen clearance of Pompe disease iPSC-derived skeletal muscle[J]. Mol Ther Methods Clin Dev, 2016, 3:16054.
[51] Yoshida S, Nakanishi C, Okada H, et al. Characteristics of induced pluripotent stem cells from clinically divergent female monozygotic twins with Danon disease[J]. J Mol Cell Cardiol, 2017, 114:234- 242.
[52] HD iPSC Consortium. Developmental alterations in Huntington’s disease neural cells and pharmacological rescue in cells and mice[J]. Nat Neurosci, 2017, 20:648- 660.
[53] Lukovic D, Artero Castro A, Delgado AB, et al. Human iPSC derived disease model of MERTK-associated retinitis pigmentosa[J]. Sci Rep, 2015, 5:12910.
[54] Ramsden CM, Nommiste B, A RL, et al. Rescue of the MERTK phagocytic defect in a human iPSC disease model using translational read-through inducing drugs[J]. Sci Rep, 2017, 7:51.
[55] Matsumoto Y, Ikeya M, Hino K, et al. New protocol to optimize iPS cells for genome analysis of fibrodysplasia ossificans progressiva[J]. Stem Cells, 2015,33:1730- 1742.
[56] Barruet E, Morales BM, Lwin W, et al. The ACVR1 R206H mutation found in fibrodysplasia ossificans progressiva increases human induced pluripotent stem cell-derived endothelial cell formation and collagen production through BMP-mediated SMAD1/5/8 signaling[J]. Stem Cell Res Ther, 2016, 7:115.
[57] Yi F, Qu J, Li M, et al. Establishment of hepatic and neural differentiation platforms of Wilson’s disease specific induced pluripotent stem cells[J]. Protein Cell, 2012, 3:855- 863.
[58] Granata A, Serrano F, Bernard WG, et al. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death[J]. Nat Genet, 2017, 49:97- 109.
[59] Park JW, Yan L, Stoddard C, et al. Recapitulating and correcting Marfan syndrome in a cellular model[J]. Int J Biol Sci, 2017, 13:588- 603.
[60] Fink JJ, Robinson TM, Germain ND, et al. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells[J]. Nat Commun, 2017, 8:15038.
[61] Francis KR, Ton AN, Xin Y, et al. Modeling Smith-Lemli-Opitz syndrome with induced pluripotent stem cells reveals a causal role for Wnt/beta-catenin defects in neuronal choles-terol synthesis phenotypes[J]. Nat Med, 2016, 22:388- 396.
[62] Ebert AD, Yu J, Rose FF Jr, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient[J]. Nature, 2009, 457:277- 280.
[63] Itzhaki I, Maizels L, Huber I, et al. Modelling the long QT syndrome with induced pluripotent stem cells[J]. Nature, 2011, 471:225- 259.
[64] Carlessi L, Fusar Poli E, Bechi G, et al. Functional and molecular defects of hiPSC-derived neurons from patients with ATM deficiency[J]. Cell Death Dis, 2014, 5:e1342.
[65] Long Y, Xu M, Li R, et al. Induced pluripotent stem cells for disease modeling and evaluation of therapeutics for Niemann-Pick disease type A[J]. Stem Cells Transl Med, 2016, 5:1644- 1655.
[66] Trilck M, Hubner R, Seibler P, et al. Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks[J]. Orphanet J Rare Dis, 2013, 8:144.
[67] Vessoni AT, Herai RH, Karpiak JV, et al. Cockayne syndrome-derived neurons display reduced synapse density and altered neural network synchrony[J]. Hum Mol Genet, 2016, 25:1271- 1280.
[68] Bell S, Peng H, Crapper L, et al. A rapid pipeline to model rare neurodevelopmental disorders with simultaneous CRISPR/Cas9 gene editing[J]. Stem Cells Transl Med, 2017, 6:886- 896.
[69] Munsat TL, Davies KE. International SMA consortium meeting[J]. Neuromuscul Disord,1992, 2:423- 428.
[70] DeRosa BA, Van Baaren JM, Dubey GK, et al. Derivation of autism spectrum disorder-specific induced pluripotent stem cells from peripheral blood mononuclear cells[J]. Neurosci Lett, 2012, 516:9- 14.
[71] Marchetto MC, Belinson H, Tian Y, et al. Altered proli-feration and networks in neural cells derived from idiopathic autistic individuals[J]. Mol Psychiatry, 2017, 22:820- 835.
[72] Ciucci F, Putignano E, Baroncelli L, et al. Insulin-like growth factor 1 (IGF- 1) mediates the effects of enriched environment (EE) on visual cortical development[J]. PLoS One, 2007, 2:e475.
[73] Cheng CM, Reinhardt RR, Lee WH, et al. Insulin-like growth factor 1 regulates developing brain glucose metabolism[J]. Proc Natl Acad Sci USA, 2000, 97:10236- 10241.
[74] Studer L, Vera E, Cornacchia D. Programming and reprogramming cellular age in the era of induced pluripotency[J]. Cell Stem Cell, 2015, 16:591- 600.
[75] Miller JD, Ganat YM, Kishinevsky S, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging[J]. Cell Stem Cell, 2013,13:691- 705.
[76] Cooper O, Seo H, Andrabi S, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease[J]. Sci Transl Med, 2012, 4:141ra90.
[77] Pearson BL, Simon JM. Identification of chemicals that mimic transcriptional changes associated with autism, brain aging and neurodegeneration[J]. Nat Commun, 2016, 7:11173.
[78] Nguyen HN, Byers B, Cord B, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress[J]. Cell Stem Cell, 2011, 8:267- 280.
[79] Vera E, Bosco N, Studer L. Generating late-onset human iPSC-based disease models by inducing neuronal age-related phenotypes through telomerase manipulation[J]. Cell Rep, 2016, 17:1184- 1192.
[80] Liu GH, Barkho BZ, Ruiz S, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome[J]. Nature, 2011, 472:221- 225.
[81] Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies[J]. Science, 2014, 345:1247125.
[82] Camp JG, Badsha F, Florio M, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocor-tex development[J]. Proc Natl Acad Sci USA, 2015, 112:15672- 15677.
[83] Otani T, Marchetto MC, Gage FH, et al. 2D and 3D stem cell models of primate cortical development identify species-specific differences in progenitor behavior contributing to brain size[J]. Cell Stem Cell, 2016, 18:467- 480.
[84] Takasato M, Er PX, Chiu HS, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis[J]. Nature, 2015, 526:564- 568.
[85] Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease[J]. Nat Cell Biol, 2016, 18:246- 254.
[86] Lee G, Ramirez CN, Kim H, et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression[J]. Nat Biotechnol, 2012, 30:1244- 1248.
[87] Choi SM, Kim Y, Shim JS, et al. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells[J]. Hepatology, 2013, 57:2458- 2468.
[88] Yamashita A, Morioka M, Kishi H, et al. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes[J]. Nature, 2014, 513:507- 511.
[89] DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: New estimates of R&D costs[J]. J Health Econ, 2016, 47:20- 33.
[90] Liang P, Lan F, Lee AS, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity[J]. Circulation, 2013, 127:1677- 1691.
[91] Kimbrel EA, Lanza R. Current status of pluripotent stem cells: moving the first therapies to the clinic[J]. Nat Rev Drug Discov, 2015, 14:681- 692.
[92] Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic[J]. Nat Rev Mol Cell Biol, 2016, 17:194- 200.
[93] Pera MF. Stem cells: The dark side of induced pluripotency[J]. Nature, 2011, 471:46- 47.
[94] Thomsen GM, Gowing G, Svendsen S, et al. The past, present and future of stem cell clinical trials for ALS[J]. Exp Neurol, 2014, 262 Pt B:127- 137.
[95] Lee AS, Tang C, Rao MS, et al. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies[J]. Nat Med, 2013, 19:998- 1004.
[96] Lee MO, Moon SH, Jeong HC, et al. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules[J]. Proc Natl Acad Sci USA, 2013, 110:E3281- 3290.
[97] Lund RJ, Narva E, Lahesmaa R. Genetic and epigenetic stability of human pluripotent stem cells[J]. Nat Rev Genet, 2012, 13:732- 744.
猜你喜欢
今日农业(2022年13期)2022-09-15 01:21:20
清华金融评论(2022年4期)2022-04-13 21:33:11
疯狂英语·新读写(2021年10期)2021-12-07 02:41:30
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:46
生物学通报(2020年10期)2020-08-13 08:52:26
奥秘(2019年8期)2019-08-28 01:47:05
知识经济·中国直销(2017年10期)2017-11-07 02:39:52
商周刊(2017年7期)2017-08-22 03:36:21
小猕猴智力画刊(2016年6期)2016-05-14 09:21:40
中国卫生(2014年2期)2014-11-12 13:00:14
