
回顾与展望:特发性间质性肺炎的临床研究
2018-05-24 05:10:56徐作军
协和医学杂志 2018年3期
徐作军
中国医学科学院 北京协和医学院 北京协和医院呼吸内科, 北京 100730
特发性间质性肺炎(idiopathic interstitial pneumonia,IIP)是一组原因不明的弥漫性非肿瘤性肺疾病,主要累及肺间质,部分患者可同时伴有肺实质、肺血管及气道受累。IIP不同病理类型临床表现和预后差异很大,治疗方案也各不相同。随着对IIP研究的不断深入,其分类和治疗方案也经历了不断演变的过程。本文对IIP分类变迁、特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)急性加重概念演变以及不同时期IPF治疗方案出台的背景进行介绍,以期为国内临床工作者更深入了解IIP的诊断和治疗进展提供参考。
1 特发性间质性肺炎的分类变迁
IIP的病理分类研究可追溯至20世纪中叶。早在1969年,著名病理学家Liebow 根据病理形态特征首次提出了IIP的分类,包括普通型间质性肺炎(usual interstitial pneumonia, UIP)、 闭塞性细支气管炎间质性肺炎(bronchiolitis obliterans interstitial pneumonia,BIP)、脱屑性间质性肺炎(desquamative interstitial pneumonia,DIP)、淋巴性间质性肺炎(lymphoid interstitial pneumonia,LIP)、巨细胞性间质性肺炎(giant cell interstitial pneumonia,GIP)5种类型[1],奠定了IIP的分类基础。随后,研究证实GIP为重金属所致职业性尘肺,故将其从此组疾病中剔除。
其后,随着对IIP研究和临床观察的不断深入,许多学者提出了不同分类方法,最具代表性的是Katzenstein于1997年提出的分类方法,包括UIP、非特异性间质性肺炎(nonspecific interstitial pneumonia,NSIP)、脱屑性间质性肺炎/呼吸性细支气管炎间质性肺病(desquamative interstitial pneumonitis/respiratory bronchiolitis interstitial lung disease,DIP/RBILD)、急性间质性肺炎(acute interstitial pneumonia, AIP)4种类型[2]。与Liebow分类相比,该分类将AIP和NSIP从UIP中分出,作为独立的类型。但Katzenstein 分类中不包括LIP和BIP, 其认为前者为淋巴组织增生性疾病,后者病变并非位于肺间质,不应归为IIP。
20世纪90年代以来,随着胸腔镜肺活检等新技术在临床的应用,对IIP的认识有了长足发展,但分类名称较混乱,不利于沟通交流。2002年美国胸科学会/欧洲呼吸学会(American Thoracic Society/European Respiratory Society,ATS/ERS)发表了关于IIP分类的国际多学科专家共识[3],此共识将IIP分为UIP、NSIP、隐源性机化性肺炎(cryptogenic organizing pneumonia,COP)、弥漫性肺泡损伤(diffuse alveolar damage,DAD)、RBILD、DIP、LIP 7种临床病理类型(表1),并对各型临床、影像学及病理特征进行了详尽描述,强调了临床-影像-病理多学科讨论是诊断IIP的“金标准”。该共识一经发表得到了业界广泛认可。
2013年ATS/ERS发表了修订的IIP分类国际多学科共识[4],该共识将IIP病理类型重新归纳,分为3大类:(1)主要的IIP;(2)罕见的IIP;(3)不能分类的IIP(表2)。其中主要的IIP,按起病轻重缓急进一步分为慢性致纤维化性间质性肺炎(包括IPF和NSIP)、急性/亚急性间质性肺炎(包括COP和AIP)以及吸烟相关性间质性肺炎(包括RBILD和DIP);罕见的IIP进一步分为特发性胸膜肺弹力纤维增生症和特发性淋巴细胞性间质性肺炎。临床曾报道过的急性球形纤维素性机化性肺炎和气道中心性间质性肺炎未被列入此次的IIP临床范畴,但被作为罕见的组织病理学类型提及。不能分类的IIP约占IIP的10%~30%,原因包括:临床、影像或病理资料不全或不一致;治疗后影像/病理类型发生变化;病理变化不能归为已知类型;多种影像或病理类型共存等。
修订的共识除对IIP病理类型进行了归纳、补充,还提出了临床分类方案,根据IIP疾病行为学特征将其分为5组,并针对不同的临床组别制定了不同治疗目标和监测策略(表3)。
2 特发性肺纤维化急性加重概念的演变
IPF是IIP中病理表现为UIP的一种慢性进行性纤维化性间质性肺病,好发于老年男性,对糖皮质激素治疗无效,中位生存期仅为2.8年,预后很差。研究表明,临床中有约4.1%的IPF患者可在疾病相对稳定的过程中突发急性加重(acute exacerbations of IPF,AE-IPF)[5],是影响IPF预后和导致患者死亡的一个重要因素。
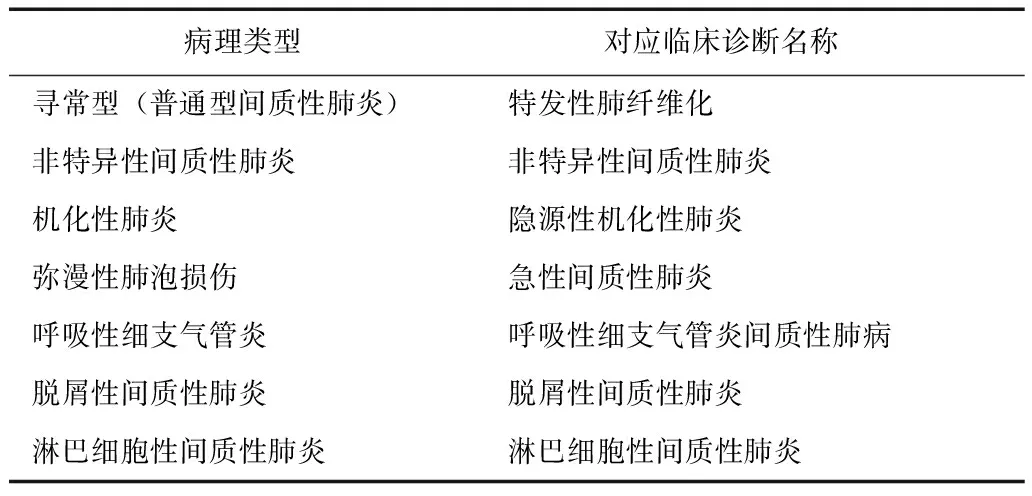
表 1 2002年美国胸科学会/欧洲呼吸学会特发性间质性肺炎 病理分型及对应临床诊断

表 2 2013年美国胸科学会/欧洲呼吸学会特发性间质性肺炎 病理分型
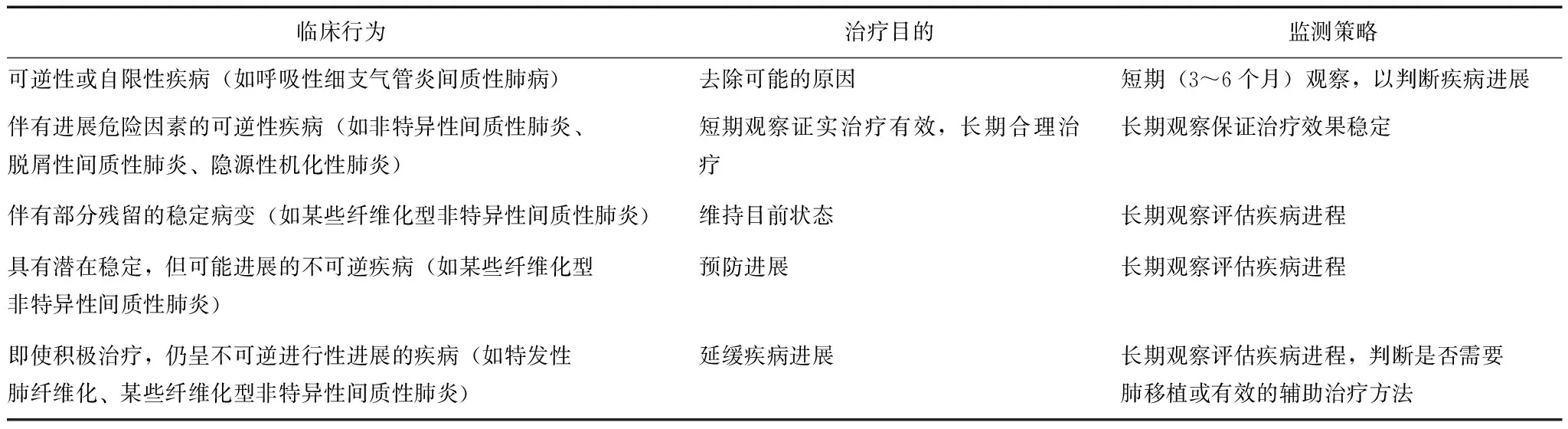
表 3 2013年美国胸科学会/欧洲呼吸学会特发性间质性肺炎临床分类国际多学科共识
1993年Kondoh等[6]最早提出了AE-IPF诊断标准:(1)近一个月内出现气促加重;(2)动脉血氧分压提示低氧血症或氧合指数<225;(3)胸部X线检查出现新的肺部浸润病灶;(4)无明显的感染或心脏疾病。但该诊断标准一直未引起临床关注,也未被大多数学者所接受。2007年美国心肺与血液研究所发起的IPFnet (the IPF Clinical Research Network)提出了AE-IPF的定义与诊断标准[7]:(1)既往或现诊断为IPF(2011年ATS/ERS标准[8]);(2)近30 d内出现无其他原因可解释的呼吸困难恶化或新出现的呼吸困难;(3)在典型的UIP型高分辨CT(high resolution CT,HRCT)表现基础上,出现新的磨玻璃影和/或实变影;(4)气管内吸引物或支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)检查无明显的肺部感染证据;(5)排除左心功能不全、肺栓塞和其他原因导致的急性肺损伤。Kondoh和IPFnet的诊断标准均将排除肺部感染列为其中之一。2016年国际AE-IPF专题工作组对AE-IPF的诊断标准行进了修订[9]:(1)既往或现诊断为IPF(2011年ATS/ERS标准[8]);(2)急性恶化或新发生的呼吸困难,典型病程1个月内;(3)UIP型HRCT表现基础上,出现新的两肺磨玻璃影和/或实变影;(4)不能以心力衰竭或液体负荷过多完全解释的恶化。
2016年国际AE-IPF专题工作组与2007年美国心肺与血液研究所的IPFnet诊断标准相比,有以下3点变化:(1)在发病时间上,强调典型病程在1个月之内;(2)不再强调排除感染因素;(3)注重AE-IPF的病理表现是DAD,而心力衰竭或液体负荷过多导致的肺水肿和呼吸困难加重不属于AE-IPF。
3 特发性间质性肺炎的治疗
在IIP中,由于IPF较为常见且预后差,故其一直是临床研究的热点。既往认为IPF是由于慢性炎症导致,治疗方法主要以糖皮质激素和免疫抑制剂为主,以致2000年ATS/ERS发表的第一个IPF国际指南中,虽然缺乏循证医学证据,仍将小剂量糖皮质激素联合免疫抑制剂(主要是环磷酰胺和硫唑嘌呤)作为IPF的推荐治疗方案[10]。此后,国际上进行了多种针对IPF的临床药物研究,其中包括N-乙酰半胱氨酸、干扰素-γ、吡非尼酮、内皮素- 1受体拮抗剂(波生坦)、抗凝药物(华法林)以及多种细胞因子拮抗剂等,但一直未得到理想结果。2011年修订的IPF国际指南,根据近10年研究结果,基本否定了前期已经进行的大多数药物对IPF的治疗作用,仅保留4种方案作为弱不推荐方案供大家选择:小剂量糖皮质激素联合免疫抑制剂(环磷酰胺或硫唑嘌呤)和N-乙酰半胱氨酸(俗称三联治疗)、单用N-乙酰半胱氨酸、吡非尼酮、抗凝治疗[8]。之后,研究者针对上述4种治疗方案又进行了多中心临床药物研究,结果否定了三联治疗、N-乙酰半胱氨酸和华法林的治疗作用。期间一种靶向治疗药物尼达尼布(为血小板衍生生长因子、成纤维细胞生长因子、血管内皮生长因子3种细胞因子络氨酸激酶受体抑制剂)的Ⅱ期和Ⅲ期临床研究显示,其可延缓IPF患者肺功能下降,故2015年第3次修订的IPF国际指南,正式推荐吡非尼酮、尼达尼布作为IPF治疗药物[11]。
对于AIP和AE-IPF,由于发病机制不清,且病情进展快,死亡高,目前国际推荐治疗方案是大剂量糖皮质激素[静脉甲基强的松龙500~1000 mg/d,3 d后改为泼尼松1 mg/(kg·d)或等效剂量激素继续治疗4~8周,根据患者病情和效果逐步减至维持剂量]联合免疫抑制剂,免疫抑制剂可选择环磷酰胺、环孢素A或他克莫司。该方案目前尚无临床试验的证据支持[11- 12],患者死亡率仍高达60%~70%,需有创机械通气患者的死亡率高达90%。
对于其他类型IIP,因多数对糖皮质激素治疗有效,故常规推荐糖皮质激素或联合免疫抑制剂治疗。
4 展望
2013年ATS/ERS发布的IIP分类国际多学科共识虽然在2002年IIP分类共识基础上作了进一步归纳、整理和补充,但仍不尽完善,比如吸烟相关性间质性肺炎,虽然成年DIP多与吸烟有关,但临床上有些DIP患者,特别是儿童并无吸烟史;另外不能分类的IIP的分类也存在一定争议。随着人们对IIP认识的不断深入,一些新的临床病理类型可能还将不断被认识和发现,因此IIP分类将是一个不断补充完善的过程。
由于不同病理类型IIP的治疗方案和预后各不相同,因此临床医生首先需要解决的问题是明确诊断。虽然IIP临床诊断要求临床-病理-放射多学科讨论,但病理学诊断居于极其重要的地位。由于多数IIP患者本身存在肺功能受损和基础疾病,加之手术肺活检易导致间质性肺病急性加重,显著限制了开胸肺活检或胸腔镜肺活检的临床应用,而经支气管镜透壁肺活检(transbronchial lung biopsy, TBLB)由于所取肺组织太小,且为盲视下随机获得,对IIP诊断意义不大。近年来,经支气管镜冷冻肺活检技术可获取足够大的肺组织,使IIP的平均诊断率达80%左右[13],明显高于传统的TBLB方法,值得今后进一步研究。
随着精准医学研究的开展,通过对基因、血清或BALF检测,寻找对IIP诊断、鉴别诊断和预后判断有意义的生物标志物已成为目前研究热点。现有研究发现,IPF患者血清中肺表面活性蛋白A(surfactant protein A,SP-A)和SP-D水平显著升高且与预后相关;NSIP患者的肺和支气管肺泡灌洗液呈现辅助T细胞1(helper T-cell 1, Th1)升高的特点,而IPF患者却呈现以趋化因子CCL- 7及其受体CCR- 7升高为特点的Th2样反应。IIP肺功能快速下降和/或生存率下降可能与血清中的某些上皮细胞或巨噬细胞相关蛋白,如涎液化糖链抗原- 6、化学趋化因子配体- 18、金属基质蛋白酶- 7等有关,这为将来通过非创伤性方法诊断和鉴别诊断IIP带来了希望,具有非常广阔的应用前景。
就治疗来说,虽然吡非尼酮和尼达尼布已被批准用于治疗IPF,但这些药物仅能够延缓肺功能下降速度,不能终止病情进展,更不能逆转病情,故远未达到临床治疗该病的要求。相信随着对IPF认识的不断深入,新的治疗药物会被逐渐开发,为IPF患者带来新的希望。
5 结语
综上所述,IIP是一组原因不明,具有不同病理类型的异原性间质性肺病,近20年来,随着人们对IIP认识的不断深入,其临床病理分类和治疗方案不断更新。随着微创介入技术的应用、分子病理学和精准医学研究的开展, IIP诊断、鉴别诊断和治疗方案的制定朝着更加精准和无创的方向发展,相信未来IIP的诊断和治疗将取得新的进展。
参 考 文 献
[1] Liebow AA, Carrington CB. The interstitial pneumonias[M]//Simon M, Potchen EJ, Lemay E. Frontiers of pulmonary radiology. New York: Grune and Stratton, 1969:102- 141.
[2] Katzenstein AL, Myers JL.Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification[J]. Am J Respir Crit Care Med, 1998,157:1301- 1315.
[3] ATS/ERS. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classifica-tion of the Idiopathic Interstitial Pneumonias[J]. Am J Respir Crit Care Med,2002, 165:277- 304.
[4] Travis WD, Costabel U, Hansell DM, et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias[J]. Am J Respir Crit Care Med, 2013, 188:733- 748.
[5] Atkins CP, Loke YK, Wilson AM. Outcomes in idiopathic pulmonary fibrosis: a meta-analysis from placebo controlled trials[J]. Respir Med,2014,108:376- 387.
[6] Kondoh Y, Taniguchi H, Kawabata Y, et al. Acute exacerbation in idiopathic pulmonary fibrosis: analysis of clinical and pathologic findings in three cases[J]. Chest, 1993, 103:1808- 1812.
[7] Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbation of idiopathic pulmonary fibrosis[J]. Am J Respir Crit Care Med, 2007, 176: 636- 643.
[8] Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management[J]. Am J Respir Crit Care Med, 2011, 183: 788- 824.
[9] Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis: an international working group report[J]. Am J Respir Crit Care Med, 2016,194:265- 275.
[10] ATS/ERS. Idiopathic pulmonary fibrosis: diagnosis and treatment international consensus statement[J]. Am J Respir Crit Care Med, 2000, 161: 646- 664.
[11] Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline[J]. Am J Respir Crit Care Med, 2015, 192:e3-e19.
[12] Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis: an international working group report[J]. Am J Respir Crit Care Med, 2016,194:265- 275.
[13] Ravaglia C, Bonifazi M, Wells AU, et al. Safety and diagnostic yield of transbronchial lung cryobiopsy in diffuse parenchymal lung diseases: A comparative study versus video-assisted thoracoscopic lung biopsy and a systematic review of the literature[J]. Respiration,2016,91:215- 227.
猜你喜欢
保健医苑(2021年7期)2021-08-13 08:47:48
昆明医科大学学报(2020年12期)2021-01-26 00:43:54
中国毕业后医学教育(2020年5期)2020-12-06 06:53:24
家庭医学(下半月)(2020年2期)2020-05-11 02:07:22
国际呼吸杂志(2019年21期)2019-11-25 09:52:20
中国中医药信息杂志(2016年7期)2016-12-01 06:07:52
中华老年多器官疾病杂志(2016年9期)2016-04-28 08:52:39
中国卫生标准管理(2015年7期)2016-01-15 03:58:41
医学研究杂志(2015年8期)2015-06-22 14:00:57
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:43
