
嵌入性金属抗癌剂的进展
2018-05-16 09:40喻欣
中国医药指南 2018年12期
喻 欣
(江汉大学医学院,湖北 武汉 430056)
1960年代,发现顺铂通过DNA交联作用,治疗卵巢癌、睾丸癌、肺癌和乳腺癌等多种癌症;但其剂量相关的肾、神经、耳毒性及抗药性(获得性或天然的)限制了它的应用[1]。在随后的开发铂类化合物中,由于不良反应和耐药性的改善,卡铂和奥沙利铂获准进入临床,但它们的抗癌活性没有显著增加。主要原因是卡铂、奥沙利铂、顺铂作用机制具有相似构效关系。以顺铂为例,Pt2+复合物含胺配体,在水合作用下解离;细胞内,氯被水取代,分子的亲电子性增加,结合亲核性核酸,并形成以共价键结合的单官能团或双官能团的加合物,DNA出现链内交联,细胞凋亡。
目前改良的方法是,将金属配合物嵌入DNA相邻的两个碱基对之间,由于此处电子少、平面芳香环多,药物可以π-π堆积或偶极-偶极相互作用,与DNA形成非共价键结合的稳定结构;DNA松弛并拉伸,最终引起细胞凋亡。过渡金属具有广泛多样的空间构型、配体以及嵌入的数目,因此在过去几十年里,出现了大量过渡金属嵌合物[2](表1)。由于过渡金属内在特性不同,嵌入配体的结合特性可以有很大区别。卟啉镍可嵌入DNA,而卟啉锌由于轴向水配体的存在则无法嵌入。本文选择过去三年间细胞毒性较高的过渡金属嵌合物,介绍它们的进展[3]。

表1 过渡金属嵌合物的结构与特性
1 铂类配合物
目前铂嵌合物中抗癌活性较高的是[Pt(HL)(AL)]2+。HL为杂环嵌入配体,包括双吡啶喹喔啉(dpq)、2,3-二甲基-dpq、苯、5,6-二甲基-苯、2,2'-联吡啶(bpy)或4,4'-二甲基-bpy。AL是一个双齿辅助配体,如S、S或R,R-1,2-二氨基环己烷(dach)。
光谱、质谱及等温滴定量热法测定发现,铂配合物(PCs)与DNA的亲和力在104~106/mol,可选择性嵌入GC之间,产生的细胞毒性高于顺铂及其类似物(在nmol浓度)。一般而言(如Pt5、Pt1),DNA的亲和力与细胞毒性正相关,AL的空间构象是影响因素,如S、S-dach(Pt1和Pt3)的细胞毒性比R、R dach(Pt1'和Pt3')高。
Pt2对细胞系的活性比顺铂高160倍(表2),转录组学研究发现其对酵母菌胞内信号分子途径的调节有明显不同于顺铂。①引起硫同化途径下调;由于这条途径中硫氧还蛋白和谷胱甘肽没有增加,可能将减少铂类化合物的耐药性。②铁和铜的转运蛋白大量增加,增强细胞毒性。
上述铂嵌合物在体内的活性有很大差异性。Pt2体外活性高,但不能抑制BD-IX大鼠腹膜种植的结肠癌细胞的生长,且可以引起肾毒性。而对移植人前列腺癌的裸鼠,Pt1的抗癌作用与顺铂作用相当,且毒性较低。
由N-[2-(氨吖啶-9-基氨基)乙基]-N-甲基乙脒组成铂抗癌物(图1),其配合物具有独特的细胞毒性,对非小细胞肺癌(NSCLC)细胞株,Pt7(图1)的IC50为5~40 nmol。其作用机制与DNA嵌入和无功能性的加合物形成有关。这种加合物对DNA的破坏性比顺铂更大:吖啶基团嵌入DNA,而铂则与邻近DNA形成加合物,复制叉停滞或DNA双链的断裂,DNA及RNA聚合酶Ⅱ被抑制,抗癌作用明显优于顺铂。
表2 Pt1-6和Pt1'-6'的细胞毒作用,以IC50(μmol)表示。IC50为72 h抑制50%细胞生长时的药物浓度(L1210:小鼠白血病细胞株;Du145:人前列腺癌细胞株;A2780:人类卵巢癌细胞株)(±s)
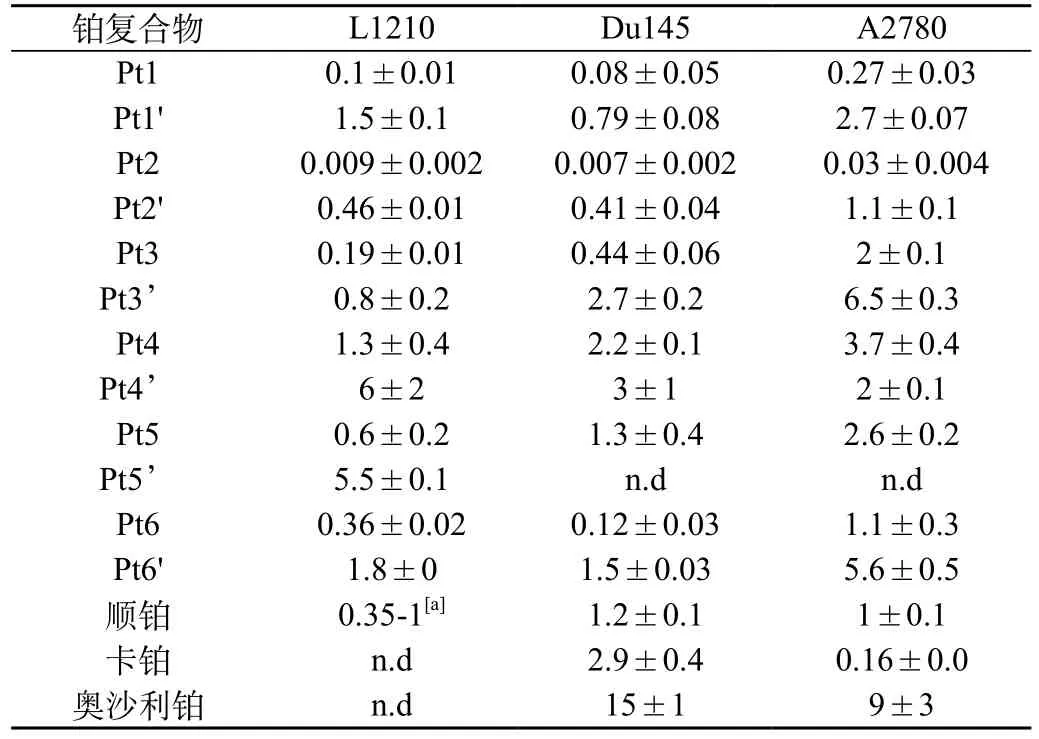
表2 Pt1-6和Pt1'-6'的细胞毒作用,以IC50(μmol)表示。IC50为72 h抑制50%细胞生长时的药物浓度(L1210:小鼠白血病细胞株;Du145:人前列腺癌细胞株;A2780:人类卵巢癌细胞株)(±s)
注:[a]引用值,.n.d=不确定
Pt1 0.1±0.01 0.08±0.05 0.27±0.03 Pt1' 1.5±0.1 0.79±0.08 2.7±0.07 Pt2 0.009±0.002 0.007±0.002 0.03±0.004 Pt2' 0.46±0.01 0.41±0.04 1.1±0.1 Pt3 0.19±0.01 0.44±0.06 2±0.1 Pt3’ 0.8±0.2 2.7±0.2 6.5±0.3 Pt4 1.3±0.4 2.2±0.1 3.7±0.4 Pt4’ 6±2 3±1 2±0.1 Pt5 0.6±0.2 1.3±0.4 2.6±0.2 Pt5’ 5.5±0.1 n.d n.d Pt6 0.36±0.02 0.12±0.03 1.1±0.3 Pt6' 1.8±0 1.5±0.03 5.6±0.5顺铂 0.35-1[a] 1.2±0.1 1±0.1卡铂 n.d 2.9±0.4 0.16±0.0奥沙利铂 n.d 15±1 9±3
铂-吖啶类物的体外细胞毒性较强,而体内实验(NCI H460移植瘤小鼠)发现,其在正常组织中的含量高于癌组织,可引发肝毒性或肾毒性。因此用苯吖啶作为配体嵌入,以增加其体积和疏水性(图1,Pt8)。Pt8的细胞毒性略小于Pt7(120~520 mmol)。
由6-苯基-2,2'-联吡啶构成的可发光的环铂化合物Pt9(图1)也具有抗癌活性。Pt9(图1)可通过嵌入DNA形成激基复合物。体外测试表明,Pt9对口腔表皮癌(KB)和神经母细胞瘤(SH-5YSY)的IC50值分别为0.009和0.010 µmol。主要抑制拓扑异构酶-DNA复合物。在植入NCI 460细胞株的裸鼠实验中,该药物可抑制60%肿瘤生长而没有不良反应。
光照激活铂卟啉Pt10(图1),可选择性定位于核内,发挥双重抗癌作用:嵌入DNA以及通过共价键结合DNA。无光照情况下,Pt10不引起DNA损伤;照射情况下(如420 nm,6.95 J/cm2),DNA出现断裂。对耐顺铂的人卵巢癌细胞株,Pt10的IC50值为0.019 µmol。
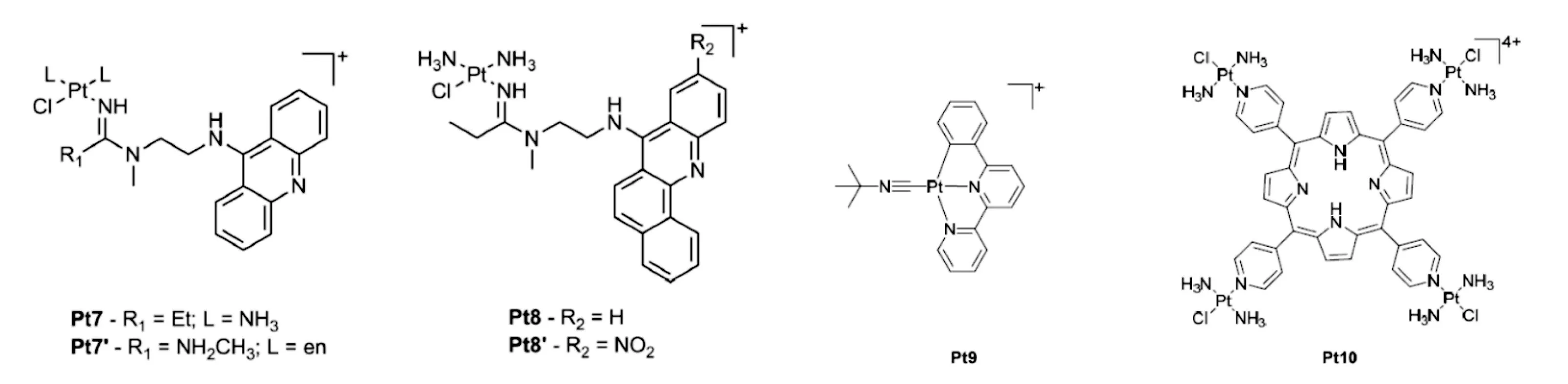
图1 Pt 7-10的结构
2 铜配合物
铜具有天然的生物利用度,癌组织增加摄取并可抑制新生血管,因而可表现出明显的抗癌活性(已有铜制剂进入Ⅱ期临床试验)。铜配合物的细胞毒作用是氧依赖性的,它嵌入DNA中并使之断裂;与相似的起始剂反应,但可以形成结构完全不同的物质。铜核酸酶[Cu(phen)2]2+可以转变成为[Cu(phen)(L)](图2),进入DNA的大沟及小沟。Cu2和Cu3(图2)对循环肿瘤DNA的亲和力约为3×107mol,是Cu1的60倍,三者均可截断质粒DNA,发挥抗SKOV3人类癌细胞的作用。
铜核酸酶还有[Cu(4phterpy)(L)2]或[Cu(4phterpy)(L)(H2O)2](L)(图2)。它们对HCT116肠癌细胞和HepG2肝癌细胞有毒性作用,但对正常成纤维细胞毒性较低。所有铜配合物均可与DNA嵌合(结合常数为105~10-6/mol),水解质粒DNA。Cu9(图2)的抗癌作用还与caspase-3和铜转运蛋白的摄取增加有关。研究还发现在苯甲酸邻位上的羟基在DNA的断裂中发挥重要作用。
铜缩氨基脲络合物包括:[Cu(Bp4mT)(µ-Cl)]2(Cu10),[Cu(µ-Bp4mT)Br]2(Cu11)(图2),[Cu(HBpT)Cl](Cu12)和[Cu(HBpT)Br](Cu13)(Bp4mT=2-苯甲酰基吡啶-4-甲基丙酮缩氨基硫脲;HBpT=2-苯并吡啶丙酮缩氨基硫脲)。铜配合物对HeLa、HepG2和nci-h460细胞的活性至少比顺铂高10倍,IC50值为(0.08±0.01)µmol。双核配合物Cu10和Cu11的活性是单核Cu12和Cu13的2倍。这些物质均可嵌入DNA并使之断裂,此外它们的抗癌作用与羟自由基和激发态氧分子也有关。
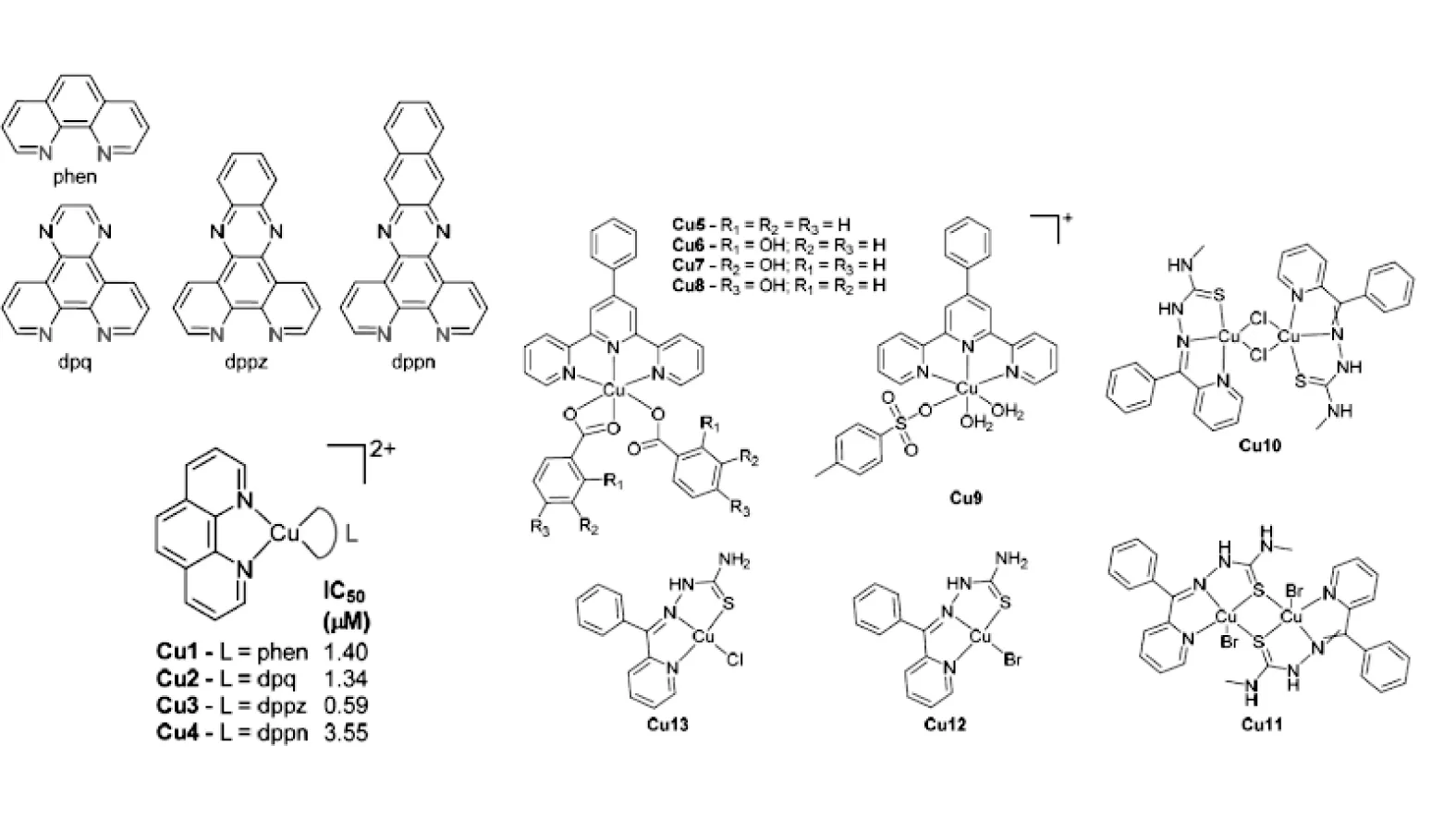
图2 Cu 1-11的化学结构
3 其他金属配合物
金配合物在药物化学领域一直比较活跃,喹喔啉环金配合物,可促进DNA嵌入,表现出细胞毒活性[Au(12,13,14,15-四氢-6,9:18,21-环亚胺[1,6]二氮杂环十八基[12,13-b]喹喔啉)]+(Au1)对人类细胞白血病和中枢神经系统肿瘤有细胞毒作用(mmol浓度)且在较高剂量时,裸鼠耐受性良好。研究表明Au1通过结合于DNA,抑制拓扑异构酶1。此外,金原子通过N-杂环卡宾与1,8-萘酰亚胺相连形成的配合物具有双重抗癌活性:DNA嵌合作用和抑制硫氧还蛋白还原酶的活性;对HT-29和MCF-7细胞株均表现出抗癌活性(mmol浓度)。
镍和锌也可作为DNA核酸酶。锌的配合物[Zn(bpbp)2]2+(bpbp=2,6-双(1-苯基-1H-苯并[d]咪唑-2-基)吡啶),在mmol浓度即表现出对各细胞系的活性,对mcf-7细胞的IC50为(2.9±0.3)µmol;该配合物嵌入DNA后使之断裂,并使磷酸化的p53基因增加,引起细胞凋亡。另一种锌配合物5-溴-8-羟基喹啉对BEL-7404和T-24的活性比顺铂高,可诱导细胞周期阻滞在细胞的G2阶段。一系列的镍异搏定缩氨基硫脲配合物亦可嵌入DNA,裂解99.8%的质粒[Ni(L)2](L=(Z)-2-(1-苄基2-氧化吲哚-3-亚甲基)-N-甲基氨基硫脲)对MCF7细胞有高度活性,IC50值的< 0.1 μmol。在H2O2存在时,镍配合物也可通过激发态氧分子等方式裂解质粒DNA。
铁(Fe2+/Fe3+)是红细胞合成、电子传递以及DNA合成必需的元素,Fe形成的配合物也是抗癌剂。二茂铁与他莫昔芬结合(Fe1),对人结肠癌(HCT-8)和急性早幼粒细胞白血病(HL-60)有细胞毒性作用,IC50值分别是0.9和1.04 µmol,但对猴的非癌性肾小球基底膜(GBM)细胞没有毒性。研究提示Fe1与双链DNA和单链DNA嵌入式结合,可使caspases3/7活化,磷脂酰丝氨酸活化,最终导致DNA碎片增加。
4 结 语
在过去的50年中,金属配合物是具有潜力的抗癌新药,1970年代,开始研究过渡金属的抗癌物。铂配合物在nmol浓度,即可嵌入DNA杀死癌细胞,发挥体内的抑瘤作用。铜的配位化合物均可以嵌入并切断DNA。镍和锌也能发挥DNA核酸酶功能。金与铁的配位化合物嵌入后,可通过不同的机制产生杀瘤作用。未来,由于过渡金属可能通过配体与特异性肿瘤的结合,扩大抗瘤谱,为临床提供更有效安全的化疗方案。
参考文献
[1] Cepeda V,Fuertes MA,Castilla J,et al.Biochemical mechanisms of cisplatin cytotoxicity[J]..Anti-Cancer Agents Med Chem,2007,7(1):3–18.
[2] Kielkopf CL,Erkkila KE,Hudson BP,et al.Structure of a photoactive rhodium complex intercalated into DNA[J].Nat Struct Mol Biol,2000,7(1):117–121.
[3] Friedman AE,Chambron JC,Sauvage JP,et al.A molecular light switch for DNA:Ru(bpy)2(dppz)2+.[J].J Am Chem Soc,1990,112(12):4960–4962.
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年6期)2021-07-31
昆明医科大学学报(2021年3期)2021-07-22
中西医结合肝病杂志(2020年2期)2020-10-27
无机化学学报(2020年7期)2020-07-20
物理化学学报(2020年4期)2020-04-24
渤海大学学报(自然科学版)(2020年3期)2020-02-02
中国中药杂志(2017年7期)2017-05-26
科技创新导报(2016年30期)2017-03-15
中国中药杂志(2016年22期)2017-02-13
