
羧甲司坦对人气道上皮细胞炎症的影响及调控机制研究
2018-05-10 01:34郭莹康健柴文戍潘殿柱车丽燕王荣李靖阎雪安晓琴
中国现代医学杂志 2018年13期
郭莹,康健,柴文戍,潘殿柱,车丽燕,王荣,李靖,阎雪,安晓琴
(1.锦州医科大学附属第一医院 呼吸科,辽宁 锦州 121001;2.中国医科大学附属第一院 呼吸疾病研究所,辽宁 沈阳 110001)
气道上皮细胞是抵御外界环境刺激物的第一道防线,吸入的香烟烟雾首先与之接触,引起气道和肺部的炎症反应,释放各种炎症介质和细胞因子。其中白细胞介素8(Interleutin-8,IL-8)是最为重要的细胞因子,是迄今发现的最强的中性粒细胞趋化因子,是中性粒细胞活化的标志。此外还有肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、IL-6、IL-10等也与炎症过程有密切关系[1]。其能有效地激活细胞内途径(即NF-κB和其他信号),导致疾病的演变和各种相关症状的产生[2]。
羧甲司坦除了作为一种临床常用的黏液调节剂[3],还具有显著的抗炎作用。研究发现,含半胱氨酸的药物,如羧甲司坦和N-乙酰半胱氨酸等能够减少促炎细胞因子(TNF-α、IL-6、IL-8)的产生[4]。已有大量的基础和临床研究证实,羧甲司坦的抗炎作用比其本身的祛痰作用更为显著。近年的临床研究发现,应用羧甲司坦可明显减少慢性阻塞性肺疾病(chronic obstructive pulmoriary disease,COPD)患者的急性加重,延长发作间歇[5]。但国外同等规模流行病学调查显示应用祛痰类药物效果不明显,分析2者之间的差异,发现国外COPD患者基础常规应用吸入性糖皮质激素,而我国PEACE研究中入选的COPD患者中仅有16.7%不规律应用吸入性糖皮质激素。由此推测合用吸入性糖皮质激素可能是差异产生的重要原因。众所周知,糖皮质激素具有全身抗炎作用,尤其是吸入糖皮质激素已被广泛应用于临床治疗哮喘及COPD患者。那么,糖皮质激素充分抗炎的“天花板效应”是否就是羧甲司坦未能发挥作用的原因呢?糖皮质激素影响羧甲司坦发挥作用的机制又是什么呢?因此,本研究旨在通过体外细胞实验证明:羧甲司坦的抗炎机制可能与糖皮质激素存在重叠,基础应用糖皮质激素者由于占用了相同的信号转导通路使某些炎症靶点受抑制,故再联合应用羧甲司坦时效果不佳。
1 材料与方法
1.1 主要试剂与仪器
羧甲司坦、布地奈德(杭州宝仑生物公司),标准研究用烟(1R3F,美国肯塔基大学),RPMI 1640培养基(美国Hyclone公司),胎牛血清、青链霉素混合液(北京Solarbio公司),EGF(美国Pepro Tech公司),PD98059(美国 Selleck公司),MTT、DMSO(美国Sigma公司),ELISA试剂盒(美国R&D公司),ERK、p-ERK、IκBα、p-IκBα抗体(英国Abcam公司),二抗(羊抗小鼠、羊抗兔)(北京中杉金桥公司),β-actin、ERK、NF-κB引物(上海生工公司),无水RNA酶(美国Thermo公司),RNAiso Plus(日本TaKaRa公司),实时聚合酶链反应(real-time PCR)试剂盒(日本TaKaRa公司),CO2细胞培养箱(美国Thermo Scientific HERA cell 150i),酶标仪(美国Bioteck公司),电泳仪(美国Bio-Rad公司),PCR仪(英国Techne公司),实时荧光定量聚合酶链反应(qRT-PCR)仪(TP800,日本TaKaRa公司)。
1.2 人气道上皮细胞培养、烟雾提取液制备及分组
1.2.1 人气道上皮细胞株(16HBE细胞) 购于中国医学科学院肿瘤细胞库。
1.2.2 烟雾提取物的制备 参照文献[6]方法,点燃一支香烟用50 ml注射器连续抽吸,香烟燃烧速度约为5 min/支,吸入的烟雾经三通管缓慢注入盛有10 ml RPMI 1640培养液的密封瓶中制成悬液,摇晃使其充分溶解,调定至pH 7.4。经0.22μm微孔滤膜过滤除菌,存于冰中。制备的(cigarette smoke extract,CSE)浓度设定为100.00%,稀释不同浓度的CSE 30 min内用于实验。
1.2.3 分组 分别给予不同干预因素,24 h后收集细胞用于实验。①对照组(N组):正常培养细胞。②模型组(CN组):细胞培养液中加入CSE。③羧甲司坦组(CC组):细胞培养液中加入CSE和10~4 M羧甲司坦。④布地奈德组(CB组):细胞培养液中加入CSE和10~8 mol/L布地奈德。⑤联合组(CCB组):先加布地奈德处理细胞1 d后再加羧甲司坦干预。⑥抑制剂组(CNP组):细胞培养液中加入CSE和100 μmol/L ERK1/2抑制剂PD98059。⑦激动剂组(CCE组):在羧甲司坦组基础上加入10 ng/ml ERK1/2激动剂EGF。
1.3 MTT法检测不同浓度CSE对细胞生存率的影响
收集对数期细胞,铺板,孵育至细胞单层铺满孔底,加入不同浓度CSE(分别为80.00%、40.00%、20.00%、10.00%、5.00%、2.50%、1.25%、0.63% 和0.00%),分别于干预后1、3、6、12、24和48 h处理细胞。每孔加入20 μl MTT溶液,继续培养4 h终止培养,每孔加入150 μl二甲基亚砜,摇床振荡10 min,酶标仪检测吸光度。终吸光度A值=A570 nm-A630 nm,细胞生存率(%)=A加药孔/A对照孔×100%。
1.4 ELISA检测炎症因子IL-8、IL-6水平的变化
准备试剂、样本,按比例稀释标准品;加样(标准品及样本)各100 μl,室温孵育2 h;吸弃,洗板4次;加酶标检测抗体200 μl,室温孵育2 h;吸弃,洗板4次;加显色底物200 μl,室温孵育30 min;加终止液50μl,450 nm波长酶标仪测量OD值,540 nm作为校正波长。
1.5 Western blot检测ERK、p-ERK、IκBα、p-IκBα蛋白水平的变化
RIPA裂解液(RIPA 75%、PMSF 10%、cocktail 15%)裂解细胞,离心取上清液采用BCA法测定蛋白浓度。与上样缓冲液混合后上样,SDS-PAGE(10%分离胶,5%浓缩胶)电泳,电转移至PVDF膜。与一抗多克隆抗体ERK、p-ERK、IκBα、p-IκBα孵育,4℃过夜。加入1︰2 500稀释的二抗室温下孵育,洗膜后显色,于凝胶自动成像分析系统下成像,采用Image J图像分析软件对条带进行灰度值测定。
1.6 qRT-PCR检测ERK、NF-κB mRNA水平的变化
Trizol法提取细胞总RNA,按cDNA合成试剂盒说明配置反应体系,在PCR仪中37℃ 15 min,85℃ 5 s逆转录合成cDNA。以此为模板分别加入NF-κB、ERK1/2、β-actin的引物(β-actin-F:5'-ATAGCACA GCCTGGATAGCAACGTAC-3';β-actin-R:5'-CACC TTCTACAATGAGCTGCGTGTG-3';ERK1/2-F:5'-CAACGTGCTCCACCGAGAT-3';ERK1/2-R:5'-TCA TGCTCAGGATCGGCAAT-3';NF-κB-F:5'-CAATCGT GCCCCCAACACT-3';NF-κB-R :5'-GTCCTCTTTC TGCACCTTGTCAC-3')和荧光染料在qRT-PCR仪上进行扩增,95℃预变性30 s,95℃变性5 s,60℃退火/延伸30 s,40个循环。基因相对表达量以2-ΔΔCt法分析。即ΔΔCt=(Ct目的基因-Ct内参基因)实验组-(Ct目的基因-Ct内参基因)对照组。各组之间的表达差异用 2-ΔΔCt表示。
1.7 统计学方法
数据分析采用SPSS 19.0统计软件,计量资料用均数±标准差(±s)表示,多组样本间比较采用单因素方差分析,组间两两比较采用LSD-t检验,P<0.05为差异有统计学意义。
2 结果
2.1 不同时间不同浓度CSE对HBE细胞生存率的影响
CSE浓度在0.63%~5.00%时,对HBE细胞的抑制作用轻微,不同时间点差异无统计学意义(P>0.05)。随着CSE浓度的升高,HBE细胞的生存率呈下降趋势。CSE浓度在10.00%~80.00%,随着作用时间的延长,细胞生存率降低,细胞大量死亡,差异有统计学意义(P<0.05)。故本实验选择5.00% CSE进行以下实验。见图1和表1。
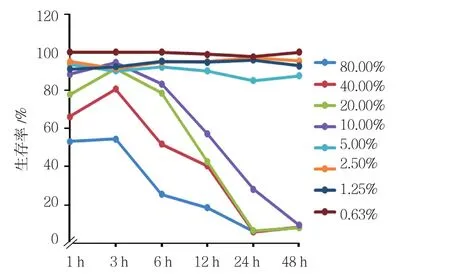
图1 不同浓度CSE对HBE细胞生存率的影响
表1 不同浓度CSE作用下不同时间HBE细胞的生存率 (%,±s)
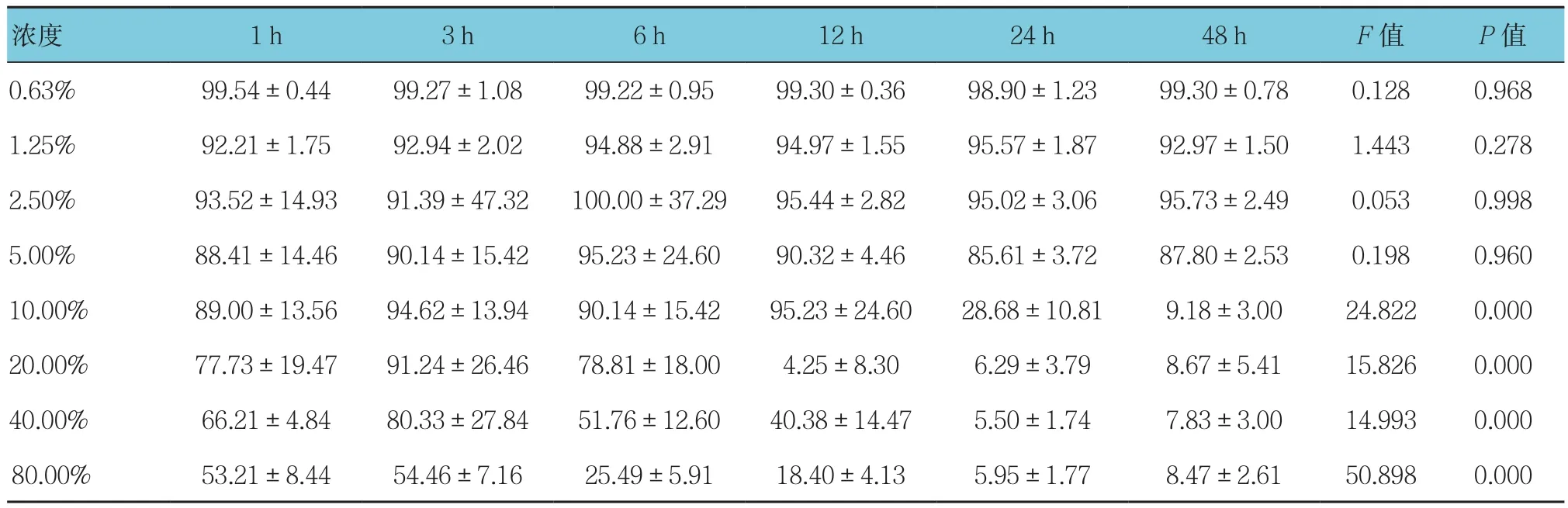
表1 不同浓度CSE作用下不同时间HBE细胞的生存率 (%,±s)
浓度 1 h 3 h 6 h 12 h 24 h 48 h F值 P值0.63% 99.54±0.44 99.27±1.08 99.22±0.95 99.30±0.36 98.90±1.23 99.30±0.78 0.128 0.968 1.25% 92.21±1.75 92.94±2.02 94.88±2.91 94.97±1.55 95.57±1.87 92.97±1.50 1.443 0.278 2.50% 93.52±14.93 91.39±47.32 100.00±37.29 95.44±2.82 95.02±3.06 95.73±2.49 0.053 0.998 5.00% 88.41±14.46 90.14±15.42 95.23±24.60 90.32±4.46 85.61±3.72 87.80±2.53 0.198 0.960 10.00% 89.00±13.56 94.62±13.94 90.14±15.42 95.23±24.60 28.68±10.81 9.18±3.00 24.822 0.000 20.00% 77.73±19.47 91.24±26.46 78.81±18.00 4.25±8.30 6.29±3.79 8.67±5.41 15.826 0.000 40.00% 66.21±4.84 80.33±27.84 51.76±12.60 40.38±14.47 5.50±1.74 7.83±3.00 14.993 0.000 80.00% 53.21±8.44 54.46±7.16 25.49±5.91 18.40±4.13 5.95±1.77 8.47±2.61 50.898 0.000
2.2 细胞上清中IL-8、IL-6水平
通过ELISA法检测细胞上清中炎症因子水平结果显示,CN组IL-8、IL-6水平均较N组升高,差异有统计学意义(P<0.05),CC组、CB组及CCB组上述指标均较模型组有所降低,差异均有统计学意义(P<0.05),但3组IL-8、IL-6水平作两两比较,差异无统计学意义(P>0.05)。见表2和图2、3。
表 2 细胞上清中 IL-8、IL-6 水平 (pg/ml,±s)
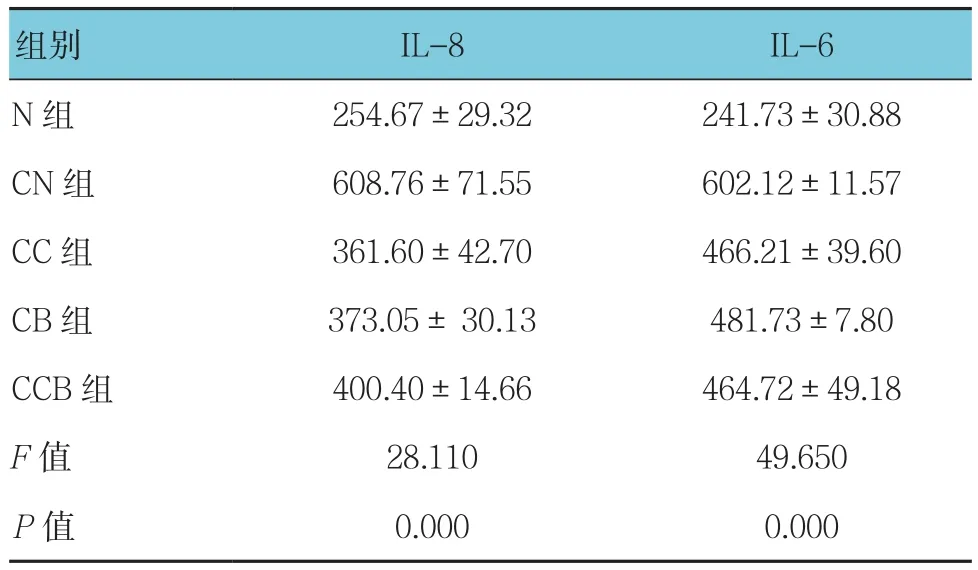
表 2 细胞上清中 IL-8、IL-6 水平 (pg/ml,±s)
组别 IL-8 IL-6 N组 254.67±29.32 241.73±30.88 CN组 608.76±71.55 602.12±11.57 CC 组 361.60±42.70 466.21±39.60 CB 组 373.05± 30.13 481.73±7.80 CCB 组 400.40±14.66 464.72±49.18 F值 28.110 49.650 P值 0.000 0.000
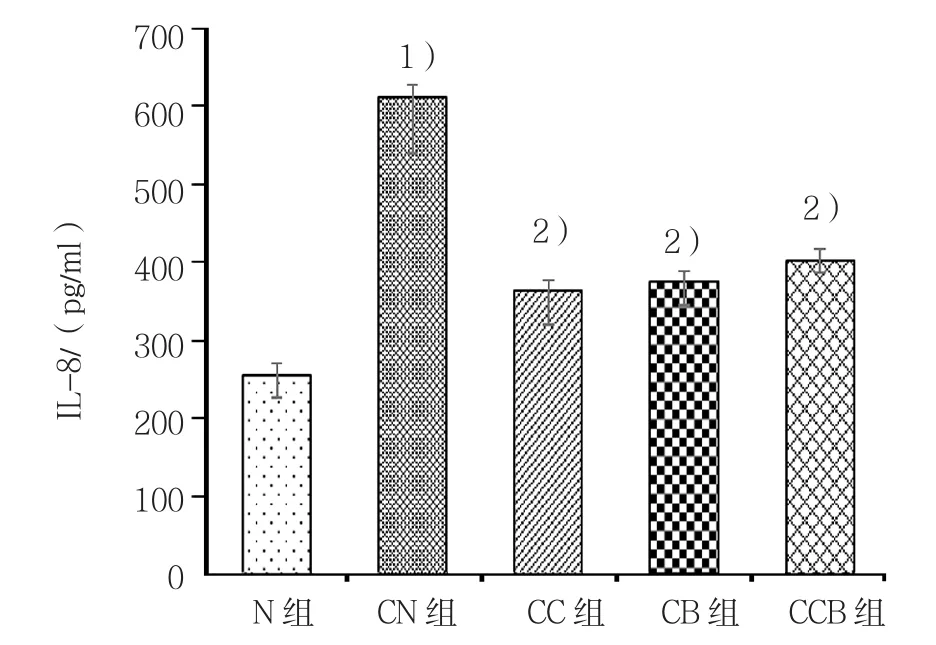
图2 细胞上清中IL-8水平
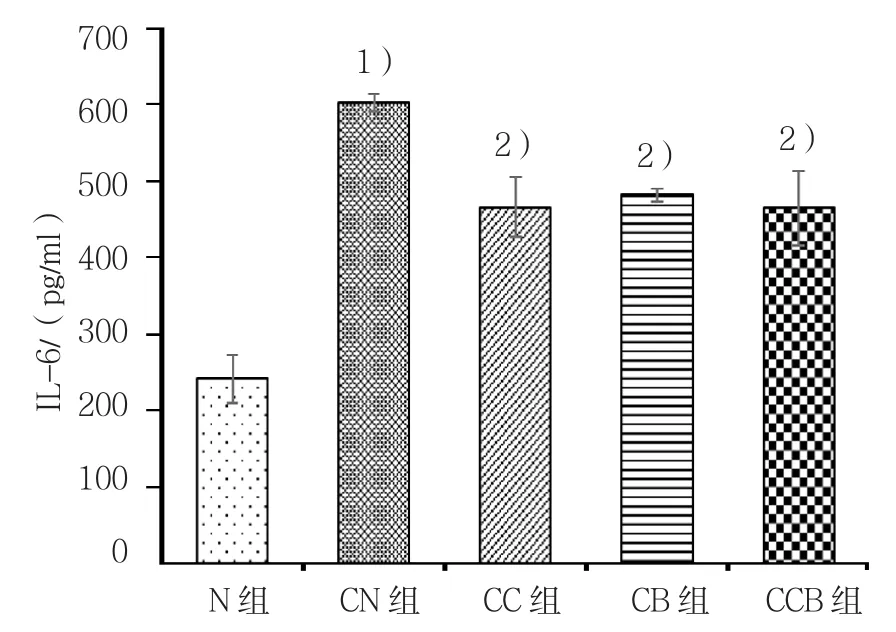
图3 细胞上清中IL-6水平
2.3 不同药物干预对HBE细胞ERK、IκBα磷酸化的影响
与N组比较,CN组HBE细胞p-ERK、p-IκBα蛋白表达均增高,差异有统计学意义(t=5.041,P=0.000);CC组、CB组及CCB组上述指标较CN组均降低,差异有统计学意义(p-ERK:3组t=3.355、2.306和 2.896,P=0.004、0.023和 0.012;p-IκBα:3组t=2.896、2.764和2.228;P=0.012、0.015和0.020);CC组与CB组P-ERK比较,差异有统计学意义(t=3.013,P=0.034),CCB组与CB组比较,差异无统计学意义(P>0.05);p-IκBα蛋白表达给药的3组间比较差异无统计学意义(P>0.05)。见图 4、5。
2.4 PD98059、EGF对HBE细胞ERK磷酸化的影响
与N组比较,CN组HBE细胞p-ERK表达增强(t=2.718,P=0.016),分别给予羧甲司坦、PD98059干预后p-ERK均降低(t=1.999和2.201,P=0.038和0.022),而给予EGF干预后,p-ERK的表达CN组与CC组比较,差异有统计学意义(t=1.812,P=0.041),CN组有所回升。见图6。
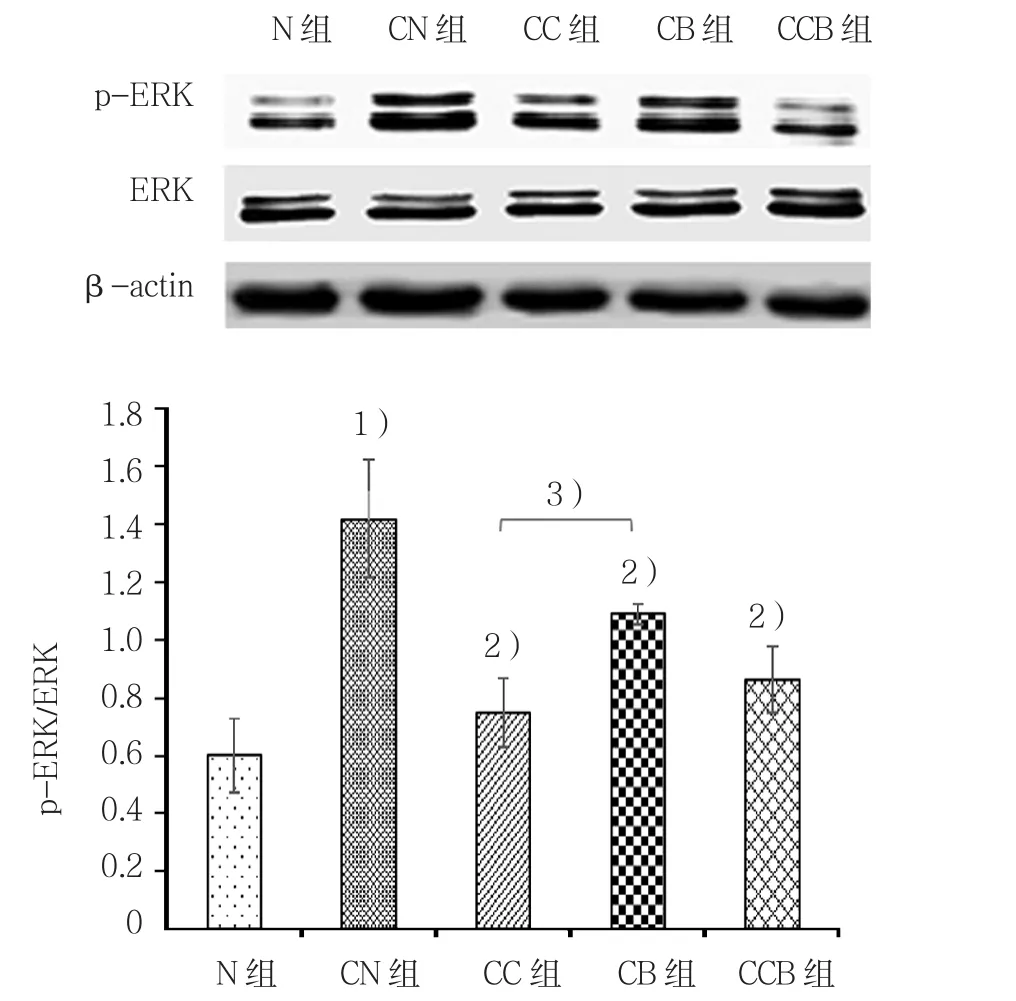
图4 p-ERK蛋白表达
与N组比较,CN组细胞ERK、NF-κB mRNA表达均增高(t=2.262和1.940,P=0.025和0.031);分
2.5 各组细胞ERK、NF-κB mRNA水平的变化
别给予羧甲司坦、布地奈德、两药联合及PD98059干预后 ERK mRNA(t=1.999、1.940、1.895和 2.896,P=0.038、0.030、0.045和0.011)及NF-κB mRNA(t=1.812、2.005、1.857 和 2.998,P=0.042、0.036、0.043和0.009)表达水平降低,而给予EGF干预后ERK和NF-κB mRNA CN组与CC组比较,差异有统计学意义(t=2.764和2.228,P=0.015和0.021),CN组有所回升。见图7、8。

图5 p-IκBα蛋白表达

图6 PD98059、EGF对ERK磷酸化的影响
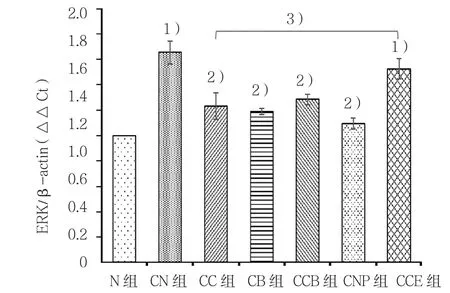
图7 ERK mRNA水平

图8 NF-κB mRNA水平
3 讨论
CSE是从香烟烟雾中提取的溶于细胞培养基的物质,其中含有丙烯醛、尼古丁、烟碱、焦油和活性氧(ROS)等成分,目前广泛用于体外实验中以研究香烟在细胞水平的作用[7-9]。本研究采用MTT法检测不同浓度CSE对HBE细胞生存率的影响,发现随着CSE浓度的升高,HBE细胞的生存率呈下降趋势。CSE浓度在0.63%~5.00%时,对HBE细胞的抑制作用轻微,不同时间差异无统计学意义。而CSE浓度在10.00%~80.00%时,随着作用时间的延长,细胞生存率降低,细胞大量死亡。
香烟烟雾中含有的大量有害物质,能够对气道和肺组织造成直接损伤,促进气道炎症,活化气道上皮细胞,产生并释放炎症介质。TNF-α、IL-8及IL-6是COPD重要的炎症介质,其来源广泛,以支气管上皮细胞和肺泡巨噬细胞产生为主,在COPD的发病机制中起重要作用。本研究结果表明,HBE细胞给予CSE干预后,其炎症因子IL-8、IL-6水平升高,而羧甲司坦、布地奈德及联合组均能不同程度降低上述炎症因子的水平,3个药物干预组间差异无统计学意义,证明CSE能够诱导炎症因子水平的升高,羧甲司坦抑制气道炎症的作用与布地奈德相近,两药联合应用未能进一步提高控制炎症的作用。
已有体外实验证明,香烟烟雾提取物(CSE)引起IL-8等炎症因子的释放与ERK和NF-κB信号转导通路的激活有关[10-12]。细胞外调节蛋白激酶(ERK)作为MAPK通路的一部分,是将信号从表面受体传导至细胞核的关键。其包括ERK1和ERK2 2个亚型,统称为ERK1/2。磷酸化激活的ERKl/2由胞质转位到核内,进而介导ELK-1、NF-κB、AP-1等转录因子的转录活化,参与细胞增殖与分化、细胞凋亡和恶变过程,并参与应激刺激、细菌产物和炎症介质等引起的生物学反应,这表明ERK通路的激活与炎症反应有着密切关系[13-15]。作为ERK通路的特异性抑制剂,PD98059通过特异性地抑制其上游激酶MEK1/2,进而抑制ERK1/2的激活与磷酸化。而EGF是研究常用的ERK通路的激动剂。本研究应用ERK通路的激动剂和抑制剂研究羧甲司坦的作用机制,结果显示CSE刺激下的HBE细胞p-ERK蛋白及ERK mRNA表达增高,分别给予羧甲司坦、布地奈德、两药联合及PD98059干预后上述指标均降低,而给予EGF干预后p-ERK及ERK mRNA较羧甲司坦组有所回升,证明羧甲司坦能够通过调控ERK的磷酸化发挥其抗炎作用。同时,本实验也观察到,糖皮质激素同样参与调节细胞内ERK-MAPK信号转导通路而抑制炎症介质,这与CLARK等[15]报道一致。
NF-κB信号转导通路是炎症反应的另1个关键环节,也是体内多条信号途径的共同交汇点。它在肺组织炎症反应进展中起着核心地位,其活化及过度表达可造成机体细胞因子失衡,产生炎症反应的级联放大效应[16-17]。NF-κB是由2个亚基组成的同源二聚体或异源二聚体,其最常见的活性形式是由P65和P50组成的异源二聚体[18]。通常状态下,NF-κB与其抑制因子IkBα螯合在细胞浆内。当机体受到内毒素、TNF-α、细菌、病毒等刺激时,IkBα迅速发生磷酸化,NF-κB被激活进入细胞核,与目标基因启动子结合并调节其转录[19]。研究发现,NF-κB的最初激活发生在肺泡巨噬细胞中,随后有活化的炎症因子进一步激化气道上皮细胞等周围细胞[21]。目前研究普遍认为NF-κB与COPD病情严重程度相关[20-23]。已有研究证实,糖皮质激素在调节NF-κB的活性中发挥了重要作用[24-25]。本研究中发现,羧甲司坦亦能降低CSE诱导的p-IκBα蛋白及NF-κB mRNA表达水平,且与布地奈德及联合组比较差异无统计学意义,说明羧甲司坦的调控作用至少部分是通过NF-κB信号转导通路进行的,这可能与糖皮质激素的作用机制存在重叠。
综上所述,羧甲司坦能够抑制香烟烟雾提取物诱导的气道炎症。羧甲司坦和布地奈德均能通过抑制ERK和NF-κB信号转导通路减轻香烟所致的气道炎症,这说明2者的作用机制存在重叠部分,因此基础应用糖皮质激素后,由于其抑制相同的炎症靶点,再联合羧甲司坦时未能体现其应有的作用,或者由于2者之间对于作用位点的竞争拮抗甚至削弱了其原本的疗效。这些尚有待于进一步的深入研究。
参 考 文 献:
[1] HENTSCHEL J, MÜLLER U, DOHT F, et al. Influences of nasal lavage collection-, processing- and storage methods on inflammatory markers - evaluation of a method for non-invasive sampling of epithelial lining fluid in cystic fibrosis and other respiratory diseases[J]. J Immunol Methods, 2014, 404(2): 41-51.
[2] RAJENDRASOZHAN S, YANG S R, EDIRISINGHE I, et al.Deacetylases and NF-kappaB in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD[J]. Antioxid Redox Signal, 2008, 10(4): 799-811.
[3] ISHIBASHI Y, TAKAYAMA G, INOUYE Y, et al. Carbocisteine normalizes the viscous property of mucus through regulation of fucosylated and sialylated sugar chain on airway mucins[J]. Eur J Pharmacol, 2010, 641(2-3): 226-228.
[4] MESSIER E M, DAY B J, KLEEBERGER S R, et al.N-Acetylcysteine protects murine alveolar type II cells from cigarette smoke injury in a nuclear erythroid 2-related factor-2-independent manner[J]. Am J Respir Cell Mol Biol, 2013, 48(5):559-567.
[5] ZHENG J P, KANG J, HUANG S G, et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease(PEACE Study): a randomised placebo-controlled study[J]. Lancet,2008, 371(9629): 2013-2018.
[6] SU Y, HAN W, GIRALDO C, et al. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells[J]. Am J Respir Cell Mol Biol, 1998, 19(5): 819-825.
[7] VAYSSIER-TAUSSAT M, CAMILLI T, ARON Y, et a1. Effects of tobacco smoke and benzo[a]pyrene on human endothelial cell and monocyte stress responses[J]. Am J Physiol Heart Cim Physiol,2001, 280(3): H1293-1300.
[8] CARNEVALI S, PETRUZZELLI S, LONGONI B, et a1. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts[J]. Am J Physiol Lung cell Mol Physiol, 2003,284(6): L955-963.
[9] VAN D T M, SMITDE VRIES M P, SLEBOS D J, et al. Cigarette smoke irreversibly modifies glutathione in airway epithelial cells[J]. Am J Physiol Lung Cell Mol Physiol, 2007, 293(5):L1156-1162.
[10] YANG S R, CHIDA A S, BAUTER M R, et al. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages[J]. Am J Physiol Lung Cell Mol Physiol, 2006,291(1): L46-57.
[11] OENEMA T A, KOLAHIAN S, NANNINGA J E, et al. Proinflammatory mechanisms of muscarinic receptor stimulation in airway smooth muscle[J]. Respir Res, 2010, 11(9): 130.
[12] BIOLLY B, VERCOUTER-EDOUART A S, HONDERMARK H, et al. FGF singnals for cell proliferation and migration through different pathways[J]. Cytokine Growth Factor Rev, 2000, 11(4):295-302.
[13] WIDMANN C, GIBOSN S, JARPE M B, et al. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human[J]. Physiol Rev, 1999, 79(1): 143-180.
[14] DZIAREKE R, JIN Y P, GUPTA D. Differential activation of extracellular signal_regulated kinase (ERK) 1, ERK2, P38 and c-Jun NH2 terminal kinase mitogen- activated protein kinases by bacterial peptidoglycan[J]. J Infect Dis, 1996, 174(4): 777-785.
[15] CLARK A R, LASA M. Crosstalk between glucocorticoids and mitogen-activated protein kinase signaling pathways[J]. Curr Opin pharmacol, 2003, 3(4): 404-411.
[16] CUZZOCREA S, CRISAFULLI C, MAZZON E, et al. Inhibition of glycogen synthase kinase-3beta attenuates the development of carrageenan-induced lung injury in mice[J]. BJ Pharmacol, 2006,149(6): 687-702.
[17] EVERHART M B, HAN W, SHERRILL T P, et al. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury J Immunol[J]. 2006, 176(8):4995-5005.
[18] KARIN M, BEN-NERIAH Y. Phosphorylation meets ubiquitination: the control of NF-κB activity[J]. Annu Rev Immunol, 2000, 18(4): 621-663.
[19] MEDURI G U, CARRATU P, FREIRE A X. Evidence of biological efficacy for prolonged glucocorticoid treatment in patients with unresolving ARDS[J]. European Respiratory Journal,2003, 42(Suppl I): I57-I64.
[20] BARNES P J, KARIN M. Nuclear factor-kappa B: a pivotal transcription factor in chronic inflammatory diseases[J]. N Engl J Med, 1997, 336(15): 1066-1071.
[21] CHAROKOPOS N, APOSTOLOPOULOS N, KALAPODI M, et al. Bronchial asthma, Chronic obstructive pulmonary disease and NF-Kb[J]. Current Medicinal Chemistry, 2009, 16(7): 867-883.
[22] CHMITZ M L, BAEUERLE P A. The p65 unit is responsible for the strong transcription activating potential of NF-Kb[J]. EMBO J.1991, 10(12): 3805-3817.
[23] CHEN L, SUN B B, WANG T, et al. Cigarette smoke enhances{beta} -defensin 2 expression in rat airways via nuclear factor-{kappa}B activation[J]. Eur Respir J, 2010, 36(3): 638-645.
[24] FEMANDES A B, ZIN W A, ROCCO P R. Corticosteroids in acute respiratory distress syndrome[J]. Braz J Med Biol Res,2005, 38(2): 147-159.
[25] LU N Z, CIDLOWSKI J A. The origin and functions of multiple human glucocorticoid receptor isoforms[J]. Ann N Y Acad Sci,2004, 1024(6): 102-123.
猜你喜欢
现代临床医学(2021年1期)2021-01-26
中华养生保健(2020年8期)2021-01-14
昆明医科大学学报(2020年11期)2020-12-28
中华养生保健(2020年7期)2020-11-16
祝您健康·文摘版(2020年7期)2020-07-13
世界最新医学信息文摘(2020年15期)2020-03-30
中国生殖健康(2019年2期)2019-08-23
世界知识(2018年19期)2018-11-21
云南中医学院学报(2015年5期)2015-07-31
中国医疗美容(2015年2期)2015-07-19
