
Cu(N3)2及其复合结构的分子动力学模拟
2018-05-03 01:51张国英王虹阳
火工品 2018年1期
张国英,王虹阳,杨 利
Cu(N3)2及其复合结构的分子动力学模拟
张国英,王虹阳,杨 利
(北京理工大学 爆炸科学与技术国家重点实验室,北京,100081)
采用Material Studio程序构建叠氮化铜Cu(N3)2、Cu(N3)2/C和Cu(N3)2/Si的超晶胞结构,以及Cu(N3)2/石墨烯的计算模型,用分子动力学方法对其力学性能进行计算。结果表明:Cu(N3)2及其复合结构各自的弹性系数区别较大,均表现出明显的各向异性,且Cu(N3)2/石墨烯的弹性系数明显不同于其它3者;复合结构Cu(N3)2/C、Cu(N3)2/Si和Cu(N3)2/石墨烯均具有良好的延展性;体积模量数值表明,加入C和Si使得Cu(N3)2断裂强度变化不明显,但石墨烯复合使得Cu(N3)2的断裂强度大幅度减小。
叠氮化铜;复合物;分子动力学;力学性能
叠氮化铜是一种起爆性能优异的起爆药,高能、绿色环保,但其感度极高,在制备和使用过程中存在潜在危险[1]。近年来,国内外研究人员对叠氮化铜的研究主要集中在获得高性能且高安全性的改性方法,其中与碳材料的复合成为了研究的热点[2]。分子动力学作为一类重要的理论计算方法,对于研究含能材料的相关性能具有非常重要的意义[3]。为改善叠氮化铜在制备和使用过程中的安全性,本文建立了Cu(N3)2、Cu(N3)2/C、Cu (N3)2/Si以及Cu(N3)2/石墨烯4种模型,采用分子动力学方法对其进行了研究,得到不同结构的力学性能参数,分析掺杂物对Cu(N3)2的力学性能的影响,以期获得更适于实际应用的Cu(N3)2复合物。
1 计算方法
1.1 力场的选择
以优化后的Cu(N3)2晶胞为初始结构,构建(2×2×4)超晶胞,如图1所示,进行分子动力学(MD)计算。采用Andersen控温和Berendersen控压,设置时间步长为1fs,总模拟步数为20万步,前10万步用于体系平衡,后10万步用于统计分析,每1 000步记录1帧轨迹数据,记录能量和温度随时间的变化。经过分子动力学模拟,体系的平衡由温度和能量的同时平衡来确定,当温度和能量在5%~10%范围内波动即可认为体系已达到平衡,如图2(a)~图2(b)所示。图2(c)~图2(b)为晶格参数及密度随时间的收敛情况。
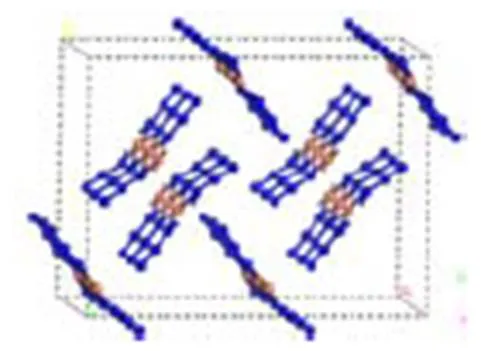
图1 Cu(N3)2超晶胞(2×2×4)结构优化
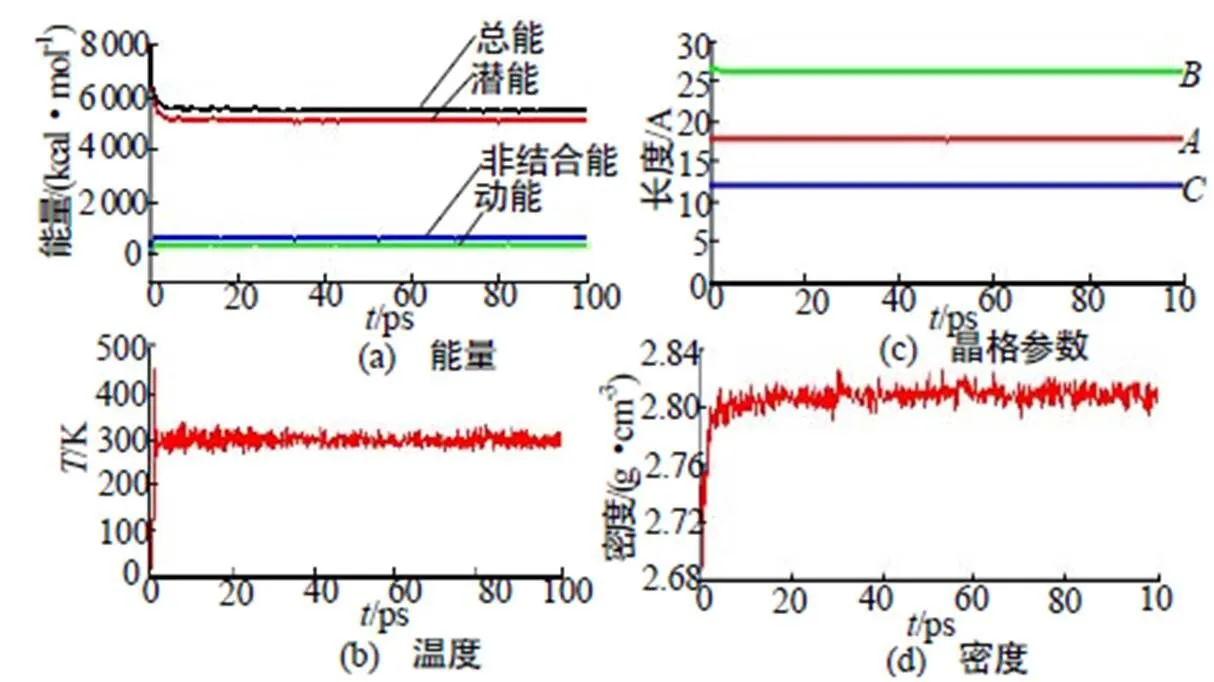
图2 298K时Cu(N3)2能量和温度的收敛情况
为了选择最佳计算力场,分别选取Universal[4]力场和Compass[5]力场,对叠氮化铜进行MD计算。通过正则系综(NVT)和等温等压系综(NPT)下的分子动力学模拟,将计算的晶格参数数据与实验值进行对比分析。计算结果与实验值的对比如表1所示。
表1 Cu(N3)2动力学模拟晶格参数及密度与实验值
Tab.1 The experimental and dynamic simulation values of lattice parameters and density of Cu(N3)2
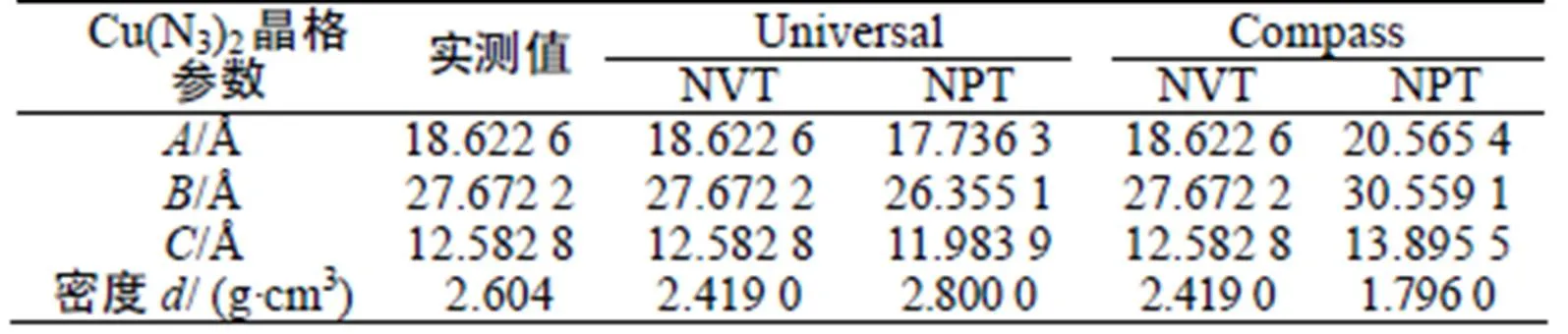
可以看出,在Universal力场下,NVT和NPT系综下MD计算的结果与实验值接近,理论计算值与实验值能较好吻合,因此Universal力场适用于叠氮化铜晶体的理论计算,均采用此力场进行模拟研究。
1.2 计算模型的构建
叠氮化铜晶体学属于正交晶系,Pbnm空间群,晶胞参数为=9.084,=13.454,=3.079,===90°。先进行Cu(N3)2单胞的结构优化,将优化后的单胞晶体与C和Si复合,得到复合结构的晶胞。优化复合结构,将复合结构单胞晶体结构进行超晶胞设置,设置超晶胞数为(2×2×4)。图3为优化后的Cu(N3)2/C和Cu(N3)2/Si超晶胞结构示意图。
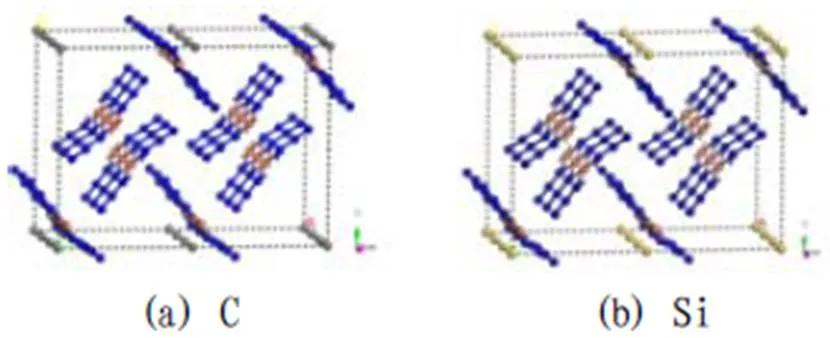
图3 Cu(N3)2与C和Si复合的超晶胞结构
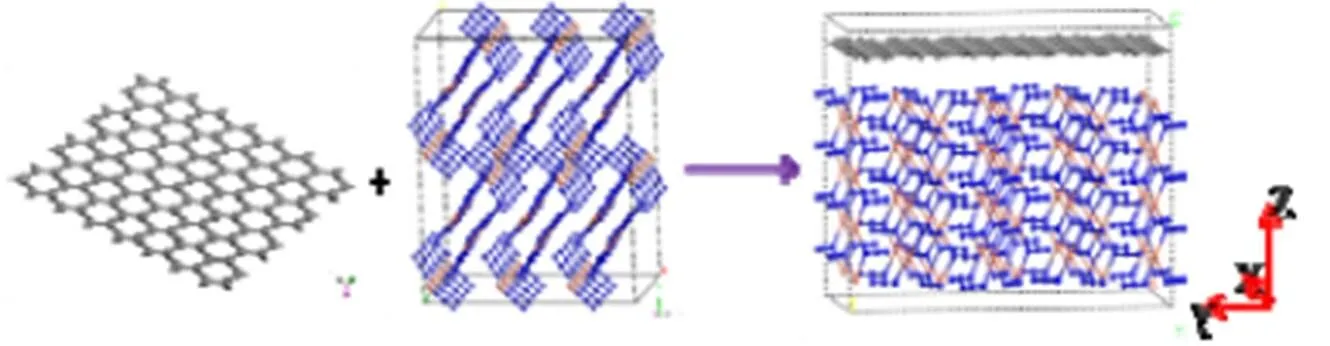
图4 Cu(N3)2与石墨烯复合结构示意图
由MS软件构建石墨烯单分子层,并使其与Cu(N3)2超晶胞复合,得到Cu(N3)2与石墨烯复合物结构,如图4所示。在Universal力场下进行MD模拟,使体系达到平衡。
1.3 力学性能模拟
用MD方法对4种计算模型进行形变量为0.5%的拉伸与纯切形变操作,得到弹性系数应变各方向应力分量C(,=1, 6)的矩阵,并计算其力学参数。
2 结果与讨论
表2为298K时4种结构的弹性系数。
表2 Cu(N3)2、Cu(N3)2/C、Cu(N3)2/Si和Cu(N3)2/石墨烯的弹性系数
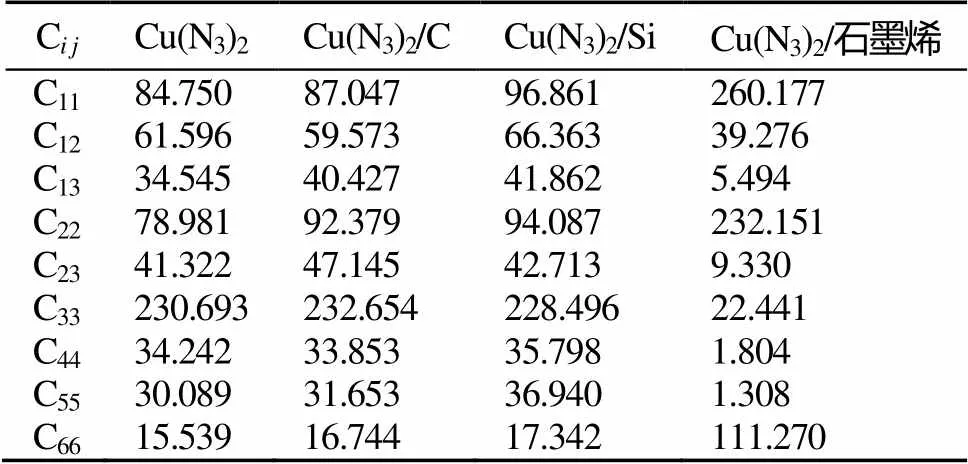
Tab.2 The elasticity coefficients of Cu(N3)2, Cu(N3)2/C, Cu(N3)2/Si and Cu(N3)2/graphene
注:C为弹性系数。
由表2可以看出,4种结构各自的弹性系数数值差别较大,均表现出明显的各向异性。略去近似等于零的系数,其中Cu(N3)2、Cu(N3)2/C和Cu(N3)2/Si的弹性系数较为接近,C33的数值最大,其次为C11、C12、C22的数值,C13、C23、C45、C55的数值较小,尤其C33的数值显著高于其它的数值。而Cu(N3)2/石墨烯复合结构的弹性系数不同,C11、C22、C66的数值显著高于其它。C、Si与Cu(N3)2复合是在不改变Cu(N3)2晶体结构的前提下,一种均匀掺杂的较为理想的复合结构,而Cu(N3)2/石墨烯的复合结构与Cu(N3)2晶体结构完全不同,是叠氮化铜简单晶面与石墨烯单分子之间的复合。计算结果表明除晶体结构、复合材料外和微观复合方式对材料的力学性能均有较大影响。表3为298K时4种结构的力学性能。
表3 Cu(N3)2、Cu(N3)2/C、Cu(N3)2/Si和Cu(N3)2/石墨烯的力学性能
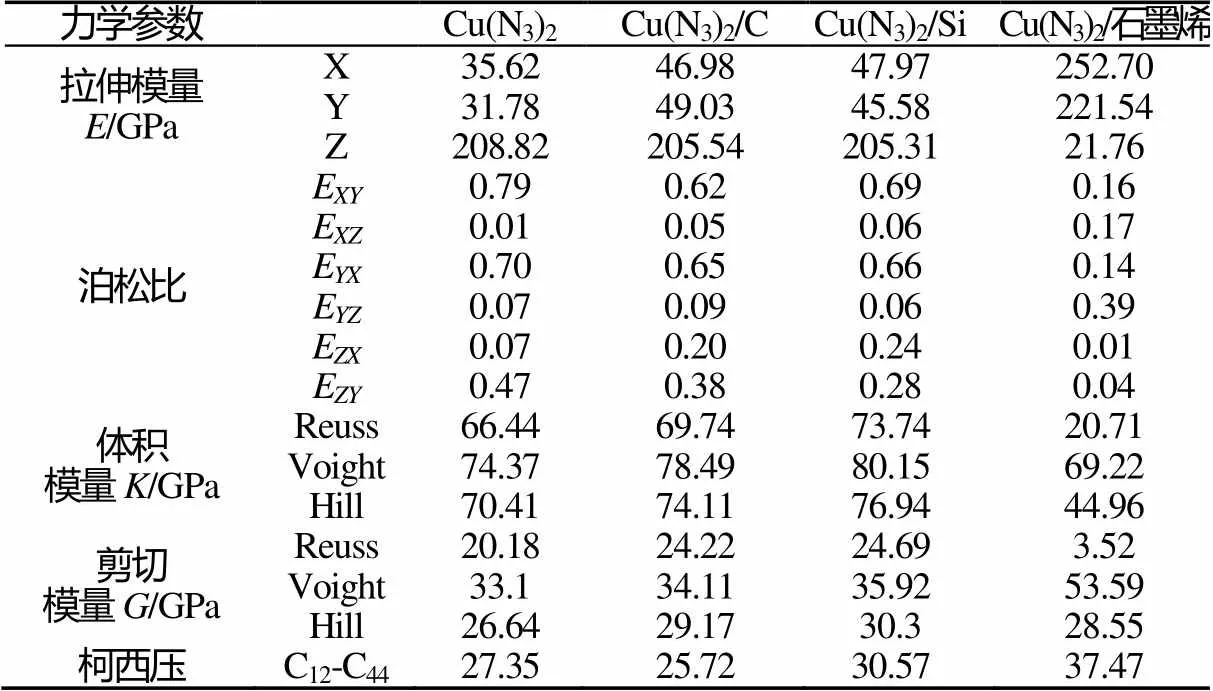
Tab.3 Mechanical properties of Cu(N3)2, Cu(N3)2/C, Cu(N3)2/Si and Cu(N3)2/Graphene
从表3中可以看出,Cu(N3)2、Cu(N3)2/C和Cu(N3)2/Si在Z方向上的拉伸模量均明显大于X、Y方向上的拉伸模量,这3种结构在Z方向上发生弹性模量变形相对较小,弹性强,材料不易变形,而在X、Y方向上弹性模量变形相对较大,材料易发生变形。对比3种计算模型,Cu(N3)2/C和Cu(N3)2/Si在X、Y方向上弹性模量比Cu(N3)2大,表明在此方向上,这两种复合结构的弹性比Cu(N3)2强,材料更易变形,在Z方向上相反。而Cu(N3)2/石墨烯与前3者不同,在X、Y方向上的拉伸模量明显大于Z方向,表明Cu(N3)2/石墨烯在X、Y方向上发生弹性模量变形相对较小,刚度大,不易变形,而在Z方向上弹性模量变形相对较大,材料易发生变形。由表3中泊松比的数值可以看出,复合结构Cu(N3)2/C、Cu(N3)2/Si和Cu(N3)2/石墨烯的值比Cu(N3)2小,表明复合结构的塑性差。柯西压(C12-C44)用来预测材料的延展性,柯西压为正值且越大,材料的延展性越高[6],延展性由大到小依次为Cu(N3)2/石墨烯、Cu(N3)2/Si、Cu(N3)2、Cu(N3)2/C。体积模量的数值表明,C和Si的加入使得Cu(N3)2的断裂强度的变化不明显,而石墨烯复合使得Cu(N3)2的断裂强度大幅度减小,有利于其实际应用。
3 结论
本研究以实测叠氮化铜晶体结构为初始结构,建立Cu(N3)2/C和Cu(N3)2/Si的超晶胞结构以及Cu(N3)2/石墨烯计算模型,结合分子动力学方法对4种模型的力学性能进行计算,得出以下结论:(1)4种结构各自的弹性系数数值区别比较大,均表现出明显的各向异性,Cu(N3)2、Cu(N3)2/C和Cu(N3)2/Si的弹性系数较为接近,而Cu(N3)2/石墨烯的弹性系数较前3者有明显的改变;(2)复合结构的泊松比较Cu(N3)2的值均较小,表明复合结构的塑性差,且Cu(N3)2/石墨烯的塑性最差, 4种结构均具有较好的延展性;(3)体积模量数值表明,C和Si的加入使得Cu(N3)2的断裂强度变化不明显,而石墨烯复合使得Cu(N3)2的断裂强度大幅度减小,有利于其实际应用。
[1] Zhou M, Li Z, Zhou Z, et al. Antistatic modification of lead styphnate and lead azide for surfactant applications[J]. Propell. Explos. Pyrot, 2013, 38(4): 569-576.
[2] Valarie Pelletier, Sayan Bhattacharyya, Isabel Knoke, Farhad Forohar, Magdy Bichay, Yury Gogotsi. Copper azide confined inside templated carbon nanotubes[J]. Adv. Funct. Mater, 2010 (20):3 168-3 174.
[3] 居学海,叶财超,徐司雨.含能材料的量子化学计算与分子动力学模拟综述[J].火炸药学报,2012, 35(2): 1-9.
[4] C. J. Casewit, K. S. Colwell, A. K. Rappii. Application of a universal force field to organic molecules[J]. J.Am.Chem. Soc., 1992(114):10 035-10 046.
[5] H. Sun. COMPASS: An ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds[J]. J. Phys. Chem. B,1998(102):7 338-7 364.
[6] D. G. Prettifor.Theoretical predicitions of structure and related properties of intermetallics[J]. Mater Sci Tech-Lond,1992, 8(4): 345-349.
Molecular Dynamics Simulation of Cu(N3)2Composites
ZHANG Guo-ying, WANG Hong-yang, YANG Li
(State Key Laboratory of Explosion Science and Technology, Beijing Institute of Technology, Beijing, 100081)
The supercell structures of copper azide (Cu(N3)2), Cu(N3)2/C, Cu(N3)2/Si and Cu(N3)2/graphene composites were established. The molecular dynamics(MD) method was used to study the changes of the mechanical property of the four samples. The MD simulation results show that the elastic coefficients of four samples are different, those of Cu(N3)2, Cu(N3)2/C and Cu(N3)2/Si are close to each other, while the elastic coefficients of Cu(N3)2/graphene is quite different. The ductility of the composite is all good. The addition of C and Si makes the change of breaking strength of Cu(N3)2not obvious, while the graphene makes the breaking strength of Cu(N3)2reduce significantly.
Copper azide;Composites;Molecular dynamics;Mechanical properties
TQ560.1
A
10.3969/j.issn.1003-1480.2018.01.013
1003-1480(2018)01-0054-03
2017-12-10
张国英(1987-),女,在读博士研究生,主要从事含能材料理论计算研究。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
昆钢科技(2022年1期)2022-04-19
纺织科学研究(2021年7期)2021-08-14
粉末冶金技术(2021年3期)2021-07-28
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
军事文摘(2020年20期)2020-11-16
军民两用技术与产品(2019年12期)2020-01-19
中学生数理化(高中版.高考理化)(2019年11期)2019-11-30
中学生数理化(高中版.高考理化)(2019年11期)2019-11-27
