
二甲双胍非降糖肾脏保护作用的研究新进展
2018-04-28 01:43刘华锋
中国药理学通报 2018年5期
薛 晶,杨 陈,安 宁,刘华锋
(广东医科大学附属医院肾病研究所,湛江市慢性肾脏病防控重点实验室,广东 湛江 524001)
目前,欧洲糖尿病研究学会、美国糖尿病协会等制定的指南均推荐二甲双胍作为2型糖尿病(type 2 diabetes mellitus, T2DM)治疗的一线用药,因其具有良好的降糖作用和安全性,是当前应用最广泛的口服降糖药。2016年,美国食品药品监督管理局又批准二甲双胍用于轻度至中度[估算肾小球滤过率(estimated glomerular filtration rate, eGFR)在30~60 mL·(min·1.73 m2)-1]肾功能不全患者[1]。二甲双胍主要通过抑制肝脏糖异生和糖原分解、减少肝脏葡萄糖输出、抑制肠道葡萄糖的吸收、促进外周靶组织对葡萄糖的摄取和利用、改善胰岛素抵抗等,发挥降糖和防治糖尿病肾病的作用,此外,近年研究发现,二甲双胍还具有独立于降糖作用之外的肾脏保护作用。
1 二甲双胍非降糖肾脏保护作用的证据
1.1二甲双胍对慢性肾脏病的保护作用众所周知,高血糖对肾脏具有明确的损伤作用,二甲双胍通过控制血糖对糖尿病肾病起肾脏保护作用已获公认,但已有研究显示,二甲双胍可直接减轻T2DM患者的尿白蛋白排泄率,改善肾脏血流动力学改变[2],与磺脲类降糖药相比,应用二甲双胍治疗T2DM合并心衰及慢性肾病的患者,其死亡率及再住院率有所降低。研究显示,与使用二甲双胍相比,口服磺脲类药物的T2DM患者发生eGFR下降、终末期肾脏病和死亡的风险增加[3]。全身和局部慢性炎症状态与慢性肾脏病密切相关,有研究发现,二甲双胍可降低患者血清中可溶性细胞间黏附分子-1、可溶性血管细胞黏附分子-1、肿瘤坏死因子α(tumor necrosis factor-α, TNF-α)等炎症介质水平,并可减少晚期氧化终产物产生,发挥抗炎和抗氧化损伤作用[4]。以上结果提示,二甲双胍对糖尿病肾病等慢性肾脏病具有独立于降糖之外的肾脏保护作用。
二甲双胍的这种作用也被大量实验研究证实,二甲双胍对肾小管上皮细胞、足细胞均有保护作用。二甲双胍可通过抗氧化应激作用,抑制高糖刺激下氧自由基介导的肾小管上皮细胞异常增殖和凋亡,从而减轻高糖对肾小管上皮细胞的损害[5];二甲双胍还可剂量依赖地抑制晚期糖基化终产物(advanced glycation end products, AGEs)的产生,并下调AGEs受体的表达[6],减少AGEs诱导的肾小管上皮细胞凋亡。持续蛋白尿不仅是慢性肾脏疾病肾小球损伤的特征,也是造成下游肾小管损伤的危险因素之一,二甲双胍对尿蛋白刺激下的肾小管起着积极的抗氧化保护作用,二甲双胍还可抑制内质网应激、抑制上皮细胞转分化和诱导自噬,减轻白蛋白所致的肾小管上皮细胞损伤[7]。足细胞损伤和丢失是糖尿病肾病早期重要的病理表现之一,二甲双胍可通过抗氧化应激作用明显减轻高糖所致的足细胞凋亡[8]。而Zhai等[9]发现,二甲双胍可上调T2DM大鼠肾脏Nephrin蛋白表达,维持足细胞正常的结构和功能,减少糖尿病大鼠的尿白蛋白排泄率。
常染色体显性多囊肾病是最常见的遗传性肾脏病,主要病理生理表现为肾小管上皮细胞异常增殖和凋亡、极性改变、细胞腔膜面持续分泌液体、囊肿进行性增大。上皮细胞顶膜上囊性纤维化跨膜转运调节因子(cystic fibrosis transmembrane regulator, CFTR)调节的氯通道的数量决定囊液的异常分泌,而哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)的异常激活则与囊泡上皮细胞的过度增生密切相关。有实验证实,二甲双胍通过腺苷酸活化蛋白激酶(AMP-activated protein kinase, AMPK)/mTOR通路阻止囊泡的增长和扩张,有效改善常染色体显性多囊肾病肾脏病变[10]。
1.2二甲双胍对急性肾损伤的保护作用二甲双胍不仅对慢性肾脏病有独立于降糖之外的潜在治疗价值,还对急性肾损伤(acute kidney injury,AKI)起到积极的防护作用。在小鼠肾脏缺血/再灌注模型,二甲双胍预处理能保持肾小管细胞膜功能的完整性,维持细胞膜上Na+,K+-ATP酶正常分布及功能,并通过直接或间接激活AMPK,增加ATP的合成,提高肾小管上皮细胞对缺血-缺氧的耐受[11]。此外,二甲双胍还可抑制大鼠肾缺血/再灌注模型TNF-α、白细胞介素6(interleukin 6, IL-6)等炎症因子的表达,减少肾小管上皮细胞凋亡,甚至减轻肾缺血/再灌注所致的肾间质纤维化等后期病变[12]。在顺铂和庆大霉素诱导的AKI研究中,二甲双胍同样可通过多种机制减少肾小管上皮细胞刷状缘脱落、凋亡、坏死、肾小管阻塞以及肾脏炎症细胞聚集,抑制血肌酐和尿素氮等生化指标的升高,促进肾脏结构和功能的恢复。
2 二甲双胍非降糖肾脏保护作用的可能机制
二甲双胍发挥降糖和非降糖作用的具体机制尚未完全阐明,目前认为,其主要通过激活AMPK发挥作用。AMPK是一种细胞能量感受器,主要参与细胞代谢,由催化亚基α和调节亚基β与γ组成。二甲双胍激活完整活细胞AMPK α亚单位第172位的苏氨酸残基,促使AMPK磷酸化,可能通过作用于AMPK上游的肝激酶B1(liver kinase B1, LKB1),激活胞内AMPK。此外,二甲双胍可阻断线粒体呼吸链复合体I的电子传递,减少ATP的合成,导致胞内AMP/ATP比例上升,从而激活AMPK。因此,二甲双胍可通过激活AMPK而发挥生物学功能。
mTOR是蛋白合成的中心调控者,在细胞生长、增殖和代谢等方面发挥重要的作用。二甲双胍激活AMPK后,可磷酸化调节mTOR信号通路的多个靶点,如激活结节性硬化复合物蛋白2、活化mTOR抑制相关蛋白Raptor等[13],最终起到抑制mTOR信号通路的作用。此外,在沉默了AMPK基因的细胞中,二甲双胍可通过抑制Rag-GTPases,从而抑制mTOR信号通路,提示二甲双胍对mTOR通路的抑制作用可部分独立于AMPK之外[14]。综上,二甲双胍可通过调节AMPK/mTOR信号通路发挥生物学功能。
2.1自噬诱导自噬是细胞将受损的细胞器或老化的长寿命蛋白包裹至双层膜囊胞,继而运送到溶酶体降解的生物学过程,自噬可将细胞内部有害物质清除,并降解出可被细胞利用的氨基酸、核酸等,因此,自噬是维持细胞内稳态的核心机制之一。二甲双胍可通过调节AMPK/mTOR通路,诱导自噬。在白蛋白所致肾小管损伤[7]、斑马鱼多囊肾[15]、缺血/再灌注[16]及顺铂所致AKI研究中发现,二甲双胍通过AMPK/mTOR诱导肾小管上皮细胞自噬,减轻细胞损伤;而抑制AMPK活性或抑制自噬诱导,则明显削弱二甲双胍的肾脏保护作用。进一步研究发现,AMPK被二甲双胍激活后,直接磷酸化自噬相关蛋白ULK1的Ser-317及717位点诱导自噬,而活化的mTOR可磷酸化ULK1的Ser-757位点,阻碍了AMPK对ULK1的上述作用而抑制自噬起始[17],二甲双胍可下调mTOR活性,解除其对ULK1的Ser-757位点的磷酸化,进而诱导自噬。因此,AMPK依赖的自噬诱导是二甲双胍发挥肾脏保护作用的重要机制之一。
2.2抗衰老生理性和应激因素所致肾脏固有细胞衰老会削弱其抗损伤和自我修复能力,增加AKI风险并加速慢性肾脏病(chronic kidney disease,CKD)进程。二甲双胍通过热量限制(caloric restriction, CR)相关方式发挥其抗衰老作用,其机制也很可能通过AMPK/mTOR信号通路发挥作用。已有研究证实,二甲双胍通过AMPK/mTOR通路,抑制高糖所致的肾小球系膜细胞[18]和近端小管上皮细胞[19]早衰及衰老的肾小管上皮细胞转分化[19]。遗憾的是,二甲双胍的抗衰老作用尚未在动物模型中证实。
2.3抗内质网应激内质网是细胞进行蛋白质折叠、翻译后修饰和运输的关键细胞器。在应激状态下,内质网功能受到干扰,引起大量蛋白质发生错误折叠或修饰,产生细胞毒性蛋白质,此即内质网应激。二甲双胍激活AMPK,诱导血红素氧合酶-1的产生,抑制衣霉素和毒胡萝卜素导致的GRP78和p-eIF2α的上调,从而抑制内质网应激,减轻肾小管上皮细胞凋亡,但沉默AMPK α1基因后,二甲双胍的上述效应只是减弱,表明其对内质网应激的抑制作用可部分独立于AMPK之外[20]。此外,蛋白超负荷通过调节ROS-c-Src激酶-mTOR通路,引起肾小管上皮细胞发生内质网应激,而应用二甲双胍可抑制白蛋白引起的内质网应激,从而减轻肾小管-间质损伤[21]。因此,二甲双胍可通过抗内质网应激,保护肾脏细胞。
2.4抗氧化应激氧化应激参与多种肾脏疾病的发生和发展。研究证实,二甲双胍对肾小管上皮细胞、足细胞均具有确切的抗氧化作用。它通过减弱反映细胞氧化水平的硫代巴比妥酸反应物(thiobarbituric acid reactive substance, TBARS)和丙二醛(malondialdehyde, MDA)等的活性,以及提高超氧化物歧化酶(superoxide dismutase, SOD)等酶类和非酶类抗氧化物水平,从而减轻庆大霉素、顺铂、肾结石等因素对肾小管上皮细胞的氧化应激损伤。还有研究发现,二甲双胍通过AMPK依赖途径,抑制NADPH氧化酶,减少线粒体活性氧簇(reactive oxygen species, ROS)产生,保护肾小管上皮细胞[5]、足细胞[8]免于高糖所致的损伤。综上,二甲双胍通过AMPK依赖的多种途径,降低细胞ROS水平,提高酶类和非酶类抗氧化物水平,发挥抗氧化应激作用。
2.5抗炎症反应核因子κB(nuclear factor kappa B, NF-κB)是一种调控机体炎症反应的重要细胞内信号,NF-κB的活化导致其下游炎症因子的产生和释放,引起炎症反应。二甲双胍剂量依赖地激活AMPK后,可抑制NF-κB抑制因子激酶(inhibitory NF-κB kinase, IKK)活性,削弱其对NF-κB抑制因子(inhibitory NF-κB, IκB)的磷酸化和降解,进而抑制NF-κB活化,降低炎症因子白细胞介素1β(interleukin 1β, IL-1β)和TNF-α的释放。二甲双胍还可通过抑制Akt、ERK信号通路,抑制NF-κB活化[22]。此外,炎症小体介导的IL-1β释放参与T2DM患者炎症反应,二甲双胍可通过AMPK依赖途径,明显抑制炎症小体介导的IL-1β分泌[23],这可能与二甲双胍诱导自噬通路后对炎症小体的下调有关[24]。此外,最新研究发现,二甲双胍也可通过下调Th17细胞和上调Treg细胞发挥抗炎作用[25]。因此,二甲双胍通过多种途径抑制肾脏炎症反应而保护肾脏。
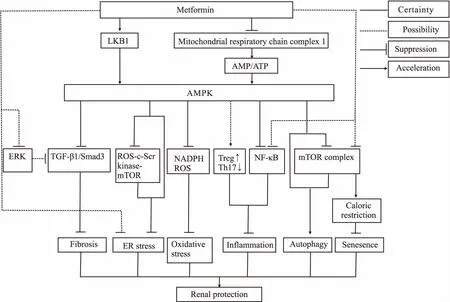
Fig 1 The underlying mechanisms of non-hypoglycemic effect of metformin in renal protection
2.6抗纤维化肾小管-间质纤维化是各类肾脏疾病发展至终末期肾衰竭的共同通路。肾小管萎缩和消失、肾间质淋巴和单核细胞浸润以及纤维组织增生是肾小管-间质纤维化最基本病理改变。转化生长因子β(transforming growth factor β, TGF-β)是促肾小管-间质纤维化的关键因子之一。在小鼠单侧输尿管梗阻模型中发现,二甲双胍通过激活AMPK,从而抑制肾小管-间质纤维化,这可能是通过抑制TGF-β1/Smad3信号通路来实现的[26]。在敲除AMPK α2基因后,二甲双胍仍有部分抑制该小鼠模型肾间质纤维化的作用[26],说明其抗肾纤维化作用并非完全通过AMPK信号通路。最近研究发现,二甲双胍也可通过竞争性抑制TGF-β1与TGF-β1受体的结合,抑制TGF-β1/Smad3信号通路[27]。此外,二甲双胍还可抑制ERK信号通路的活化,抑制TGF-β信号通路[28],这可能与其阻断ERK信号通路,影响TGF-β介导的Smad3的转录活性有关[29]。因此,二甲双胍通过AMPK依赖及非依赖途径抑制TGF-β1/Smad3通路介导的肾间质纤维化,延缓肾脏纤维化进程。
3 总结与展望
近年来大量研究发现,二甲双胍通过AMPK依赖和非依赖途径诱导自噬、抗衰老、抗氧化应激、抗内质网应激、抗炎症、抗纤维化等,发挥非降糖的肾脏保护作用(Fig 1)。虽然二甲双胍非降糖肾脏保护作用仍有待高质量的临床试验进一步验证,但其具有开发成防治CKD和AKI药物的巨大潜力,有望成为“新用老药”的典范。
参考文献:
[1] U.S.Food and Drug Administration.FDA Drug Safety Communication: FDA revises warnings regarding use of the diabetes medicine metformin in certain patients with reduced kidney function[EB/OL].(2016-04-08)[2017-07-10].https://www.fda.gov/drugs/drugsafety/ucm493244.htm.
[2] Amador-Licona N, Gu Zar-Mendoza J, Vargas E, et al.The short-term effect of a switch from glybenclamide to metformin on blood pressure and microalbuminuria in patients with type 2 diabetes mellitus[J].ArchMedRes, 2000,31(6):571-5.
[3] Hung A M, Roumie C L, Greevy R A, et al.Comparative effectiveness of incident oral antidiabetic drugs on kidney function[J].KidneyInt, 2012,81(7):698-706.
[4] Chakraborty A, Chowdhury S, Bhattacharyya M.Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients[J].DiabetesResClinPract, 2011,93(1):56-62.
[5] Takiyama Y, Harumi T, Watanabe J, et al.Tubular injury in a rat model of type 2 diabetes is prevented by metformin[J].Diabetes, 2011,60(3):981-92.
[6] 李业琼,叶山东,翟丽敏,等.二甲双胍对2型糖尿病模型大鼠肾组织AGEs表达的影响[J].中国药理学通报, 2016,32(5):703-7.
[6] Li Y Q,Ye S D,Zhai L M,et al.Effects of metformin on expression of renal tissue AGEs in type 2 diabetic rats[J].ChinPharmacolBull, 2016,32(5):703-7.
[7] Allouch S, Munusamy S.Metformin attenuates albumin-induced alterations in renal tubular cellsinvitro[J].JCellPhysiol, 2017,232(12):3652-63.
[8] Piwkowska A, Rogacka D, Jankowski M, et al.Metformin induces suppression of NAD(P)H oxidase activity in podocytes[J].BiochemBiophysResCommun, 2010,393(2):268-73.
[9] Zhai L, Gu J, Yang D, et al.Metformin ameliorates podocyte damage by restoring renal tissue nephrin expression in type 2 diabetic rats[J].JDiabetes, 2017,9(5):510-7.
[10] Takiar V, Nishio S, Seo-Mayer P, et al.Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis[J].ProcNatlAcadSciUSA, 2011,108(6):2462-7.
[11] Seomayer P W, Thulin G, Zhang L, et al.Preactivation of AMPK by metformin may ameliorate the epithelial cell damage caused by renal ischemia.[J].AmJPhysiolRenalPhysiol, 2011,301(6):F1346-57.
[12] Wang M, Weng X, Guo J, et al.Metformin alleviated EMT and fibrosis after renal ischemia-reperfusion injury in rats[J].RenFail, 2016,38(4):614-21.
[13] Kimura N, Tokunaga C, Dalal S, et al.A possible linkage between AMP- activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway[J].GenesCells, 2003,8(1):65-79.
[14] Kalender A, Selvaraj A, Kim S Y, et al.Metformin, independent of AMPK, inhibits mTORC1 in a Rag GTPase-dependent manner[J].CellMetab, 2010,11(5):390-401.
[15] Chang M Y, Ma T L, Hung C C, et al.Metformin inhibits cyst formation in a Zebrafish model of polycystin-2 deficiency[J].SciRep, 2017,7(1):7161.
[16] Declèves A, Sharma K, Satriano J.Beneficial effects of AMP-activated protein kinase agonists in kidney ischemia-reperfusion: autophagy and cellular stress markers[J].NephronExpNephrol, 2015,128(3-4):98-110.
[17] Kim J, Kundu M, Viollet B, et al.AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1[J].NatCellBiol, 2011,13(2):132-41.
[18] Guo Y, Wang J, Cai G, et al.AMPK-mediated downregulation of connexin43 and premature senescence of mesangial cells under high-glucose conditions[J].ExpGerontol, 2014,51:71- 81.
[19] Dong D, Cai G Y, Ning Y C, et al.Alleviation of senescence and epithelial-mesenchymal transition in aging kidney by short-term caloric restriction and caloric restriction mimetics via modulation of AMPK/mTOR signaling[J].Oncotarget, 2017,8(10):16109-21.
[20] Kim H, Moon S Y, Kim J, et al.Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis[J].AmJPhysiolRenalPhysiol, 2015,308(3):F226-36.
[21] Lee E K, Jeong J U, Chang J W, et al.Activation of AMP-activated protein kinase inhibits albumin-induced endoplasmic reticulum stress and apoptosis through inhibition of reactive oxygen species.[J].NephronExpNephrol, 2012,121(1-2):e38-48.
[22] Isoda K, Young J L, Zirlik A, et al.Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells[J].ArteriosclerThrombVascBiol, 2006,26(3):611-7.
[23] Lee H M, Kim J J, Kim H J, et al.Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes[J].Diabetes, 2013,62(1):194-204.
[24] Cho M H, Cho K, Kang H J, et al.Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome[J].Autophagy, 2014,10(10): 1761-75.
[25] Lee S, Lee S H, Yang E, et al.Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between Th17/Treg balance[J].PLoSOne, 2015,10(9):e0135858.
[26] Feng Y, Wang S, Zhang Y, et al.Metformin attenuates renal fibrosis in both AMPKα2-dependent and independent manners[J].ClinExpPharmacolPhysiol, 2017,44(6): 648-55.
[27] Xiao H, Zhang J, Xu Z, et al.Metformin is a novel suppressor for transforming growth factor (TGF)-beta1[J].SciRep, 2016,6:28597.
[28] Shen Y, Miao N, Xu J, et al.Metformin prevents renal fibrosis in mice with unilateral ureteral obstruction and inhibits Ang II-induced ECM production in renal fibroblasts[J].IntJMolSci, 2016,17(2):E146.
[29] 张彦伟,张志强,吴丽贤,等.雷公藤内酯醇减少放射性肺纤维化中肌成纤维细胞活化与抑制TGF-β1/ERK/Smad3通路相关[J].中国药理学通报, 2017,33(5):630-6.
[29] Zhang Y W, Zhang Z Q, Wu L X, et al.TPL’s suppression on activation of myofibroblasts in radiation induced lung fibrosis related to its inhibition on TGF-β1/ERK/Smad3 pathway[J].ChinPharmacolBull, 2017,33(5):630-6.
猜你喜欢
肝博士(2022年3期)2022-06-30
现代临床医学(2021年1期)2021-01-26
山东医药(2021年28期)2021-01-11
健康之家(2020年15期)2020-05-08
中成药(2019年12期)2020-01-04
中国医学创新(2019年9期)2019-08-19
中成药(2017年10期)2017-11-16
滨州医学院学报(2016年2期)2016-05-27
医学研究杂志(2015年12期)2015-06-10
癌变·畸变·突变(2015年3期)2015-02-27
