
CXCL12/CXCR4在佐剂性关节炎大鼠脾脏中的表达及芍药苷-6′-O-苯磺酸酯的作用
2018-04-28 01:43:40陈镜宇
中国药理学通报 2018年5期
高 梅,章 敏,司 敏,陈镜宇,魏 伟
(安徽医科大学临床药理研究所,抗炎免疫药理学教育部重点实验室,抗炎免疫药物安徽省协同创新中心,安徽 合肥 230032)
芍药苷-6′-O-苯磺酸酯(CP-25)是课题组对白芍总苷(total glucosides of paeony,TGP)的主要有效成分芍药苷(paeoniflorin,Pae)结构进行酯化修饰,合成的新型活性单体。研究表明,TGP和Pae均具有抗炎免疫调节作用,可明显抑制关节炎大鼠的关节肿胀,改善关节、脾脏和肠系膜淋巴结病理变化。然而,Pae生物利用度低,导致其起效慢。课题组前期研究表明,CP-25(25、50、100 mg·kg-1)对佐剂性关节炎(adjuvant-induced arthritis,AA)大鼠有确切的治疗作用,不仅可以明显减轻AA大鼠关节炎症,改善滑膜细胞异常增殖[1];还具有免疫调节作用,可以调节树突状细胞的功能[2],改善AA大鼠脾脏的病理[3],包括减轻白髓增生和红髓充血,减少动脉周围淋巴鞘细胞密度,减轻淋巴小结、边缘区和生发中心增生,但具体机制不清楚。外周淋巴组织淋巴细胞选择性的游走及归巢主要由趋化因子介导。趋化因子CXCL12及其受体CXCR4通过介导T、B细胞的迁移、增殖和活化,参与类风湿关节炎(rheumatoid arthritis,RA)的免疫反应[4]。RA病人的关节滑膜、滑液和血液中CXCL12的水平明显高于正常人[5],但RA时脾脏中CXCL12/CXCR4的表达情况,以及脾脏的病理改变是否与CXCL12/CXCR4有关,仍不清楚。
本研究在前期研究的基础上,成功建立AA模型,采用CP-25的有效浓度(50 mg·kg-1)给药,观察脾脏组织的病理结构改变、脾脏中CXCL12和CXCR4的表达,探讨AA脾脏病理改变与CXCL12/CXCR4的关系,以及CP-25免疫调节作用的机制。
1 材料与方法
1.1材料
1.1.1实验动物 Sprague-Dawley(SD)大鼠,♂,体质量(180±20)g,清洁级,购于安徽医科大学实验动物中心,实验动物生产许可证号:SCXK(皖)2011-002。
1.1.2药物与试剂 CP-25由本所药化室提供,白色结晶状粉末,纯度大于98%;卡介苗(Bacillus Calmette-Guerin,BCG)购自北京生物制品所;CXCL12 ELISA试剂盒购于武汉华美公司;CXCL12、CXCR4抗体购于Abcam公司;ECL发光显色试剂盒购于Thermo Scientific 公司。
1.1.3仪器 Olympus显微成像系统;ImageQuant LAS 4000 荧光及化学发光成像系统(美国 GE 公司);BioTek Elx×808 酶标仪(美国BioTek公司);RM2135石蜡切片机(德国莱卡)。
1.2方法
1.2.1AA模型的建立 将80℃水浴灭活1 h的BCG与高压灭菌的石蜡充分研磨混匀,制成 10 g·L-1的CFA,于每只大鼠右后足跖皮内注射0.1 mL致炎,造模当天设为d 0。将造模成功的大鼠随机分为模型组、CP-25(50 mg·kg-1)组、MTX(0.5 mg·kg-1)组,同时设置正常对照组。造模后d 14开始给药,d 28处死大鼠。CP-25按照50 mg·kg-1体质量灌胃给药,每天1次;MTX按照0.5 mg·kg-1体质量灌胃给药,每3 d一次。
1.2.2脾脏病理学观察 造模后d 28,分别将正常对照组、模型组、CP-25组和MTX组大鼠放血处死,迅速剪开大鼠腹部,分离、留取脾组织,剪取部分脾组织置于4%甲醛溶液中固定,不同浓度梯度乙醇逐级脱水,石蜡包埋,制作病理切片(厚约5 μm),经HE染色后,观察脾脏组织病理改变,进行病理评分。脾脏病理从动脉周围淋巴鞘(periarteriolar lymphoid sheath,PALS)的细胞密度、淋巴小结(lymphoid nodule,LN)增生进行判定,按照病变从轻到重,进行0~3级病理评分。PALS细胞密度评分:0=正常;1=细胞密度轻微增大;2=细胞密度中度增大,有细胞拥挤;3=细胞密度重度增大,有细胞叠加。LN增生评分:0=正常;1=轻微增生,可见生发中心;2=中度增生,生发中心明显;3=过度增生,生发中心多见。
1.2.3免疫组化法检测脾脏组织中CXCL12和CXCR4水平 脾脏组织制作病理切片后,经脱蜡水化,细胞通透,抗原修复,稀释相应比例的CXCL12和CXCR4的一抗,4℃过夜,二抗孵育,显色剂显色后封片。采集400倍视野图像,利用Image-Pro Plus分别对各切片中免疫组化染色阳性表达进行半定量分析,测定脾脏组织中CXCL12和CXCR4蛋白染色平均光密度(mean optical density,MOD)值后,进行统计学分析。
1.2.4Western blot检测脾脏组织中CXCR4水平 剪取部分脾组织,常规裂解(RIPA ∶PMSF=99 ∶1)充分研磨后,Lowry 法蛋白定量,加蛋白上样缓冲液,沸水煮8 min,取15 μL蛋白样品上样,进行SDS-PAGE电泳,转至PVDF膜上,脱脂奶粉/TPBS封闭2 h,放入稀释相应比例的CXCR4一抗,4℃过夜,加入稀释的二抗,室温孵育2 h。ECL试剂盒显影。结果采用凝胶成像系统扫描后,以特异性条带浓度与面积的乘积为有效值,反映蛋白表达水平。
1.2.5ELISA检测脾脏组织中CXCL12水平 剪取各组大鼠脾组织0.15 g,分别加PBS 100 μL,充分研磨后,离心取上清,用ELISA试剂盒测CXCL12的表达。
2 结果
2.1CP-25对AA大鼠脾脏病理的影响Fig 1 HE染色结果显示,正常对照组大鼠脾脏结构清晰,红髓与白髓界限较明显;与正常对照组相比,AA组大鼠红髓与白髓界限模糊,红髓充血,白髓增生,PALS的细胞密度增加,LN增多,CP-25可以减少PALS的细胞密度,改善LN增生。
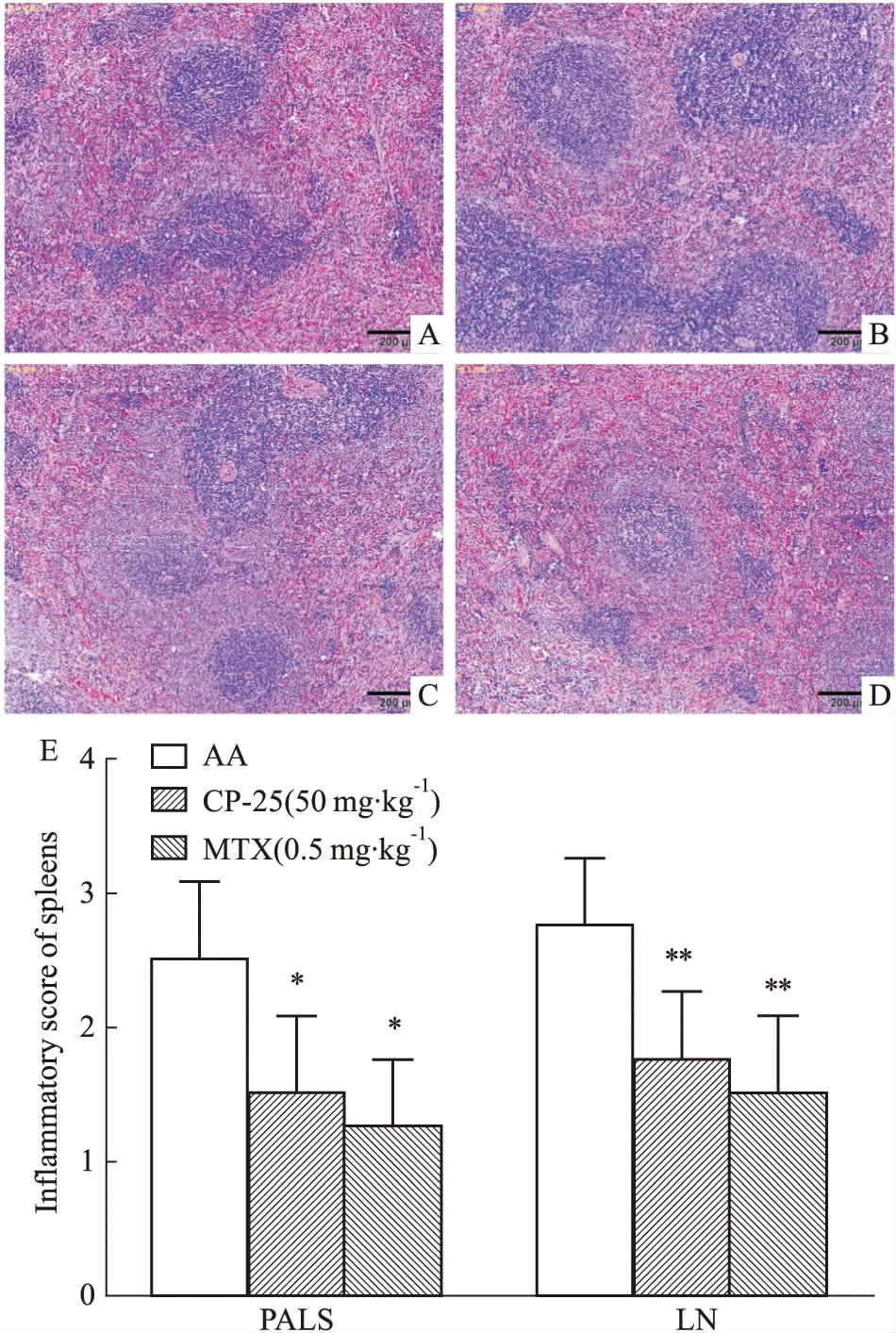
Fig 1 Effect of CP-25 on histopathology of spleen in AA rats(×100)
2.2CP-25对AA大鼠脾脏CXCL12表达的影响ELISA结果显示,与正常对照组相比,AA组大鼠脾脏中CXCL12的表达明显升高(P<0.05);与AA组相比,CP-25组和MTX组中CXCL12表达下降(P<0.05)(Fig 2A)。免疫组化结果显示,各组脾脏淋巴细胞胞质均呈免疫阳性反应;与正常对照组比较,AA组大鼠脾脏中CXCL12表达增强(P<0.05);与AA组比较,CP-25组和MTX组中CXCL12表达减弱(P<0.05)(Fig 2B~2F)。
2.3CP-25对AA大鼠脾脏CXCR4表达的影响Western blot结果显示,AA大鼠脾脏中CXCR4表达较正常对照组增加(P<0.01),CP-25和MTX可以降低脾脏中CXCR4的表达(P<0.05)(Fig 3A)。免疫组化结果显示,各组脾脏淋巴细胞胞质均呈免疫阳性反应;与正常对照组比较,AA组大鼠脾脏中CXCR4表达增强(P<0.05);与AA组比较,CP-25组和MTX组中CXCR4表达减弱(P<0.05)(Fig 3B~3F)。
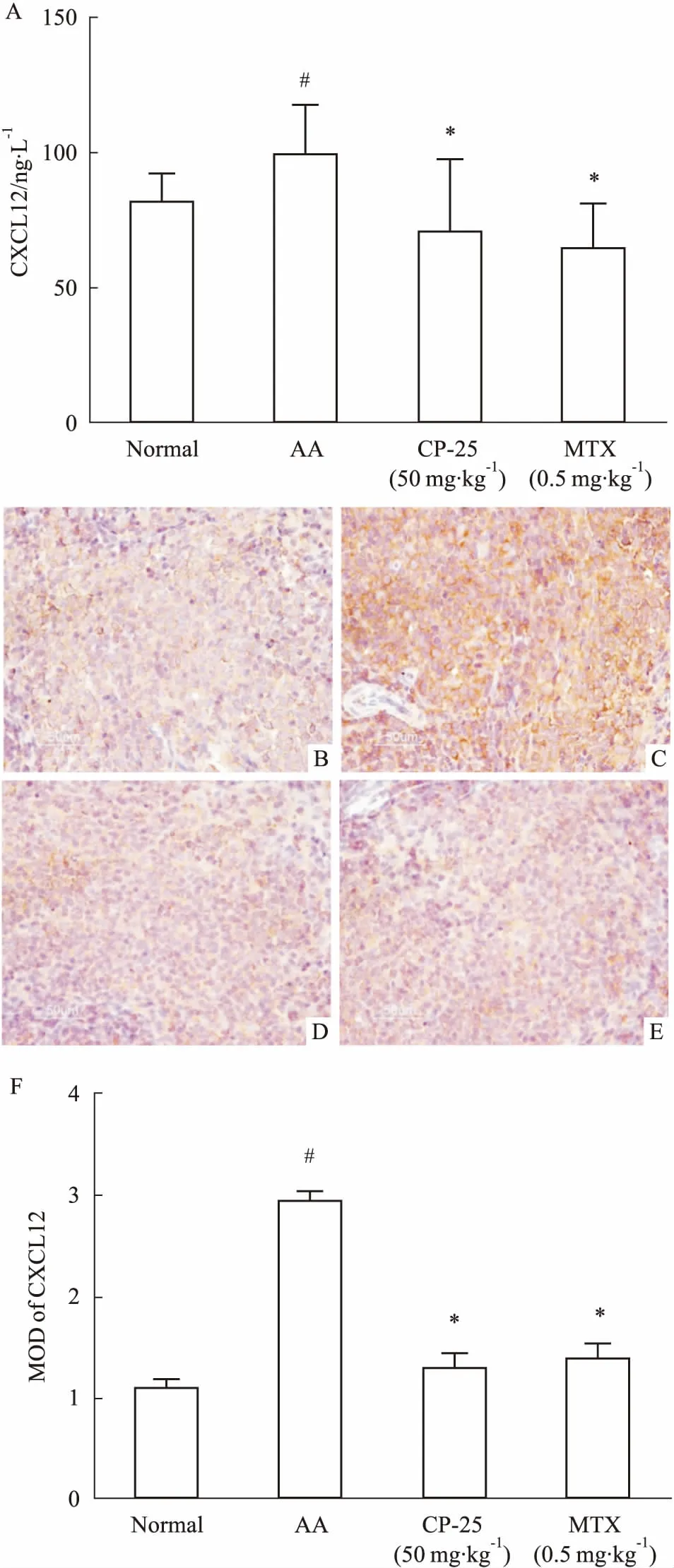
Fig 2 Effect of CP-25 on expression of CXCL12 in spleen of AA
A: ELISA results of CXCL12; B: Normal; C: AA; D: CP-25 50 mg·kg-1; E: MTX 0.5 mg·kg-1; F:MOD of CXCL12.#P<0.05vsnormal;*P<0.05vsAA.
2.4脾脏病理评分与CXCL12/CXCR4的相关性相关性分析结果显示,脾脏组织的PALS和LN增生的病理评分与脾脏组织中CXCL12和CXCR4的表达之间均存在正相关(Fig 4)。CP-25改善脾脏病理改变可能与下调脾脏中CXCL12/CXCR4的表达有关。
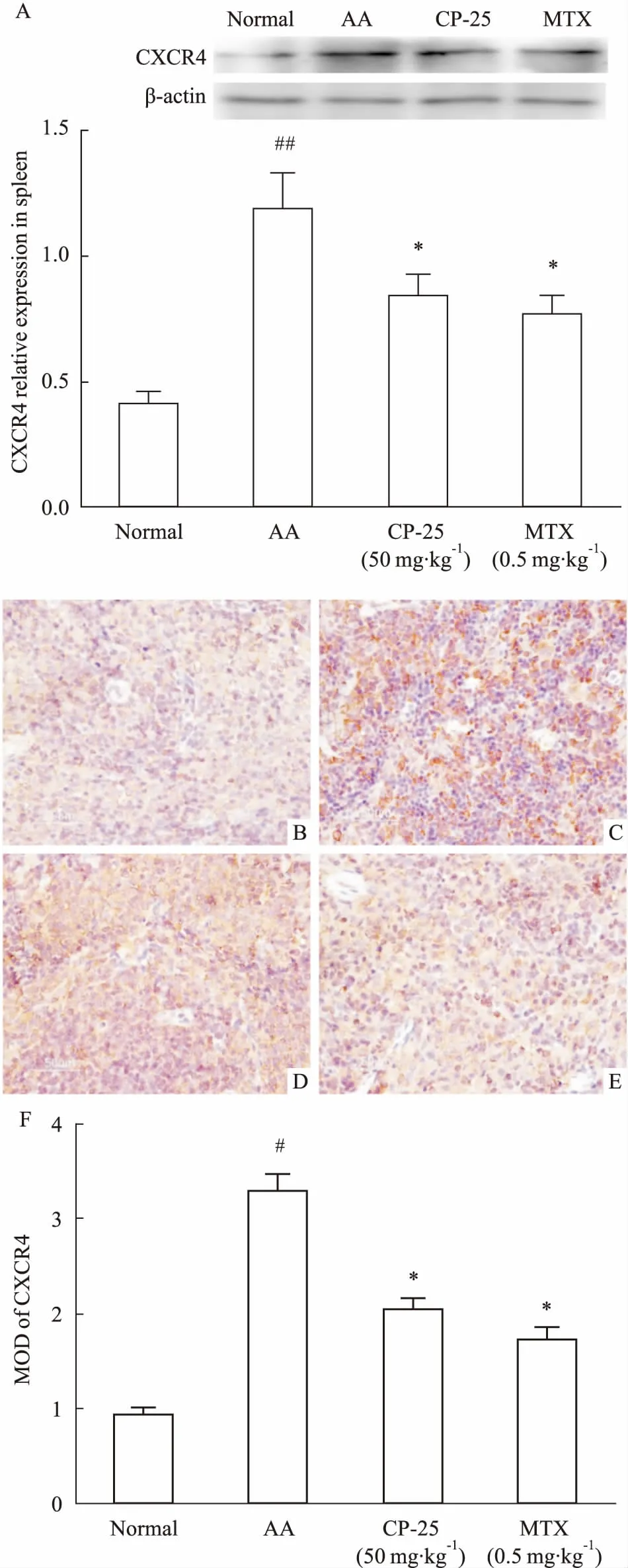
Fig 3 Effect of CP-25 on expression of CXCR4 in spleen of AA rats
3 讨论
AA模型是一种T细胞依赖的免疫性炎症动物模型,在临床表现和病理机制等方面与RA有许多相似特征,是研究RA病理机制的较理想动物模型[6]。脾脏是人体最大的免疫器官,正常脾脏由白髓和红髓组成,白髓是淋巴细胞的聚集区,由PALS和LN组成。红白髓交界处的边缘区富含T、B淋巴细胞,是抗原提呈、启动免疫应答的场所。PALS主要由大量的T细胞构成,RA时大量T细胞异常活化,增殖分化为效应T细胞,这些T细胞浸润在滑膜组织和关节液,产生大量的促炎细胞因子,导致滑膜炎症和关节破坏[7]。淋巴小结主要由大量B细胞构成,是B细胞活化和分化为浆细胞、记忆B细胞的场所。活化的B细胞通过抗原提呈参与T细胞的活化,通过产生自身抗体和记忆B 细胞维持RA自身免疫反应的持续存在[8]。病理结果显示,AA模型大鼠脾脏白髓与红髓界限模糊,白髓增生,动脉周围淋巴鞘细胞密度增加,淋巴小结增生,提示AA时脾脏中T、B细胞聚集,发生免疫应答。
CXCL12属于趋化因子家族,又称为基质细胞衍生因子1(stromal cell-derived factor-1,SDF-1),具有多种生物功能,如干细胞动员、炎性细胞浸润和血管生成,参与各种疾病过程,如癌症、艾滋病、自身免疫性疾病和缺血性疾病等[9]。CXCL12有两种受体,即CXCR4和CXCR7,主要通过CXCR4发挥作用,CXCR4是7次跨膜G蛋白偶联受体[10]。在RA的滑膜组织及关节液中CXCL12的水平增加,CXCL12可以促进血管形成[11],介导表达CXCR4的T细胞和B细胞向滑膜组织迁移[4]。CXCL12促进软骨细胞分泌MMP-1、MMP-13,使软骨降解,使用CXCR4受体拮抗剂AMD3100可以降低滑膜液中MMP-1和MMP-13的水平[12],改善胶原诱导关节炎(collagen-induced arthritis,CIA)小鼠的全身表现。这些研究表明,CXCL12/CXCR4有利于炎性效应细胞向关节迁移并且滞留,促进新生血管形成,从而推动炎性过程发展,在RA的发病机制中发挥重要作用。但CXCL12/CXCR4是否与RA的脾脏病理改变有关仍不清楚。本实验结果显示,AA大鼠脾脏PALS细胞密度和LN增生,均与CXCL12以及CXCR4的表达水平存在正相关,提示CXCL12/CXCR4与AA脾脏的病理改变有关,可能通过趋化作用,促进淋巴细胞在脾脏的聚集,其具体的作用及机制有待进一步研究。
CP-25是课题组对TGP的主要有效成分Pae进行结构修饰,合成的新型活性单体。研究表明,Pae能明显抑制 CIA大鼠B淋巴细胞增殖,降低血清中IgA、IgG和IgM水平,降低血清中B细胞刺激因子和脾脏BAFF-R表达[13]。Pae抑制关节炎模型大鼠滑膜细胞增殖,降低IL-1、TNF-α和PGE2等促炎性细胞因子分泌[14]。然而Pae生物利用度低,导致其起效慢。前期研究发现,CP-25 (25、50、100 mg·kg-1) 对AA大鼠具有明显的抗炎和免疫调节作用,可以减轻AA大鼠的关节肿胀,改善关节、脾脏病理变化,减少巨噬细胞活化,减少血清中促炎细胞因子如IL-1β、IL-6、IL-17、TNF-α的水平,上调抗炎细胞因子TGF-β1的水平[3]。CP-25通过调节PGE2-EP4-cAMP和TNF-α-TNFR1-TRADD-TRAF2-NF-κB信号通路,调节树突细胞功能[2]。CP-25可以抑制滑膜细胞的异常增殖,下调滑膜细胞分泌的IL-1β、IL-6和TNF-α的水平[1]。本实验发现,AA大鼠给予CP-25或者MTX后,CXCL12和CXCR4表达减少,脾脏PALS的细胞密度减少,LN增生得到改善,且脾脏的这些病理改变与CXCL12/CXCR4的表达呈正相关,提示CP-25可能通过减少AA大鼠脾脏组织中CXCL12和CXCR4的表达,减少淋巴细胞向脾脏的迁移,从而改善脾脏的病理改变,减轻免疫反应,发挥免疫调节作用。MTX作为RA的治疗药物,可以降低炎性因子如TNF-α、IL-6、IL-10、MMP-3等水平,但由于免疫抑制作用,产生一些副作用,如骨髓抑制、胃肠道反应、皮疹、过敏性肺炎等[15]。在本实验中,MTX作为阳性对照药的作用与CP-25相似,但在实验过程中出现老鼠体质量下降、食欲不振、缺乏运动等现象,而CP-25给药组未出现此现象,这也与前期课题组研究结果相符,表明CP-25的耐受性好。因此,作为一种有前景的治疗RA的药物,不良反应少可能是CP-25的优势。
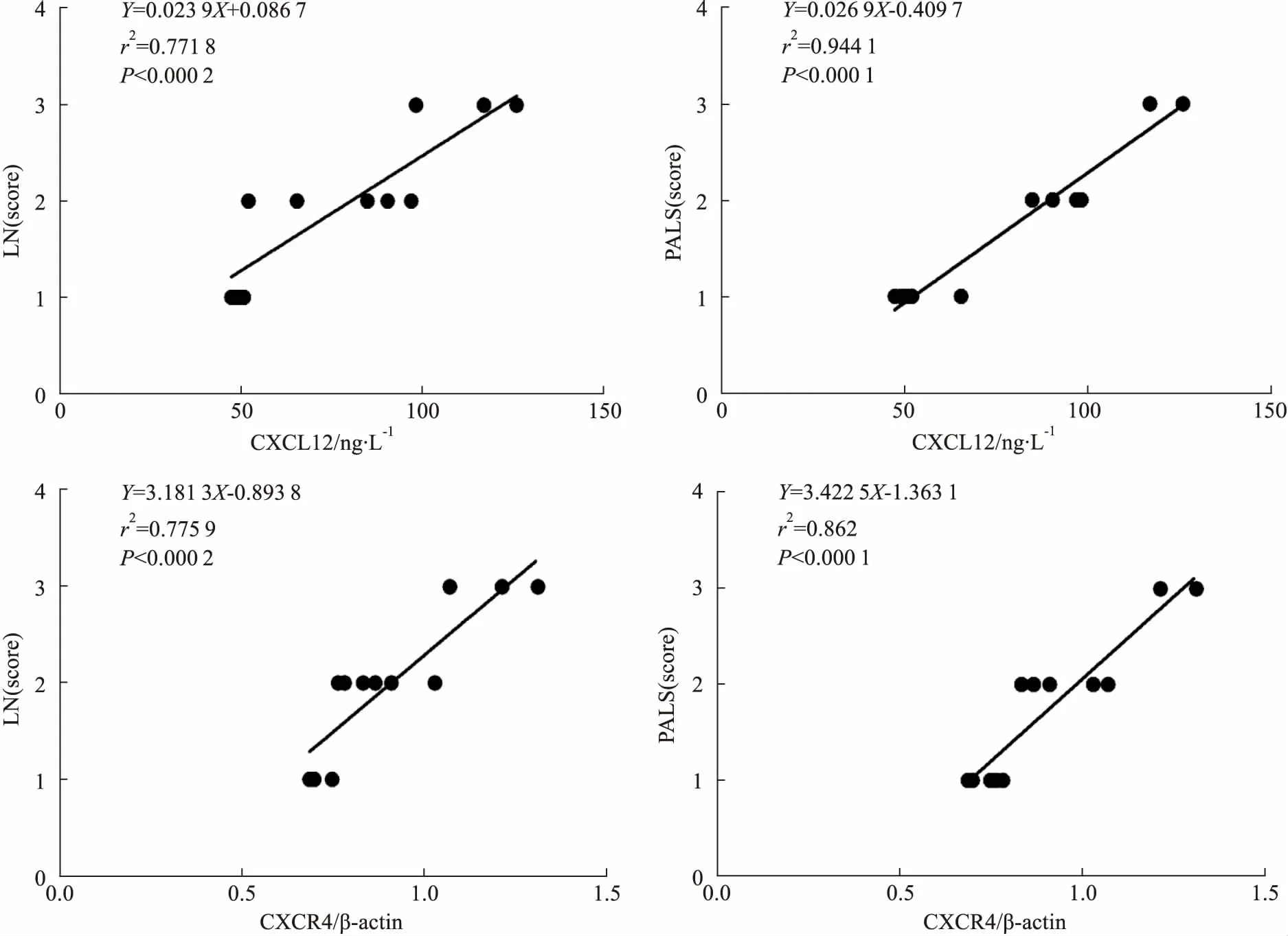
Fig 4 Correlation between pathological changes of spleen and expression of CXCL12/ CXCR4
综上所述,CXCL12及其受体CXCR4的高表达与AA脾脏的病理改变有关,可能是通过趋化作用促进淋巴细胞在脾脏的聚集,从而促进AA脾脏中免疫反应的发生;抑制AA大鼠脾脏中CXCL12和CXCR4的表达可能是CP-25发挥免疫调节作用的机制之一。
参考文献:
[1] Jia X,Wei F,Sun X,et al.CP-25 attenuates the inflammatory response of fibroblast-like synoviocytes co-cultured with BAFF-activated CD4(+) T cells[J].JEthnopharmacol,2016,189:194-201.
[2] Li Y,Sheng K,Chen J,et al.Regulation of PGE2 signaling pathways and TNF-alpha signaling pathways on the function of bone marrow-derived dendritic cells and the effects of CP-25[J].EurJPharmacol,2015,769:8-21.
[3] Chang Y,Jia X,Wei F,et al.CP-25,a novel compound,protects against autoimmune arthritis by modulating immune mediators of inflammation and bone damage[J].SciRep,2016,6:26239.
[4] Nanki T,Takada K,Komano Y,et al.Chemokine receptor expression and functional effects of chemokines on B cells: implication in the pathogenesis of rheumatoid arthritis[J].ArthritisResTher,2009,11(5):R149.
[5] Hansen I B,Ellingsen T,Hornung N,et al.Plasma level of CXC-chemokine CXCL12 is increased in rheumatoid arthritis and is independent of disease activity and methotrexate treatment[J].JRheumatol,2006,33(9):1754-9.
[6] 孙晓静,徐 澍,贾晓益,等.大鼠佐剂性关节炎病程不同阶段T细胞亚群及重要器官的病理学变化[J].中国药理学通报,2015,31(4):475-81.
[6] Sun X J,Xu S,Jia X Y,et al.Variation of T-cell subsets in spleen and tissue histopathological changes in rats with adjuvant arthritis[J].ChinPharmacolBull,2015,31(4):475-81.
[7] Mellado M,Martínez-Muoz L,Cascio G,et al.T cell migration in rheumatoid arthritis[J].FrontImmunol,2015,6:384.
[8] Nevius E,Gomes A C,Pereira J P.Inflammatory cell migration in rheumatoid arthritis: a comprehensive review[J].ClinRevAllergyImmunol,2016,51(1):59-78.
[9] Peled A,Wald O,Burger J.Development of novel CXCR4-based therapeutics[J].ExpertOpinInvestigDrugs,2012,21(3):341-53.
[10] Villalvilla A,Gomez R,Roman-Blas J A,et al.SDF-1 signaling: a promising target in rheumatic diseases[J].ExpertOpinTherTargetsm, 2014,18(9):1077-87.
[11] Janssens R,Mortier A,Boff D,et al.Truncation of CXCL12 by CD26 reduces its CXC chemokine receptor 4- and atypical chemokine receptor 3-dependent activity on endothelial cells and lymphocytes[J].BiochemPharmacol,2017,132:92-101.
[12] Wei F,Moore D C,Wei L,et al.Attenuation of osteoarthritis via blockade of the SDF-1/CXCR4 signaling pathway[J].ArthritisResTher,2012,14(4):R177.
[13] Li P P,Liu D D,Liu Y J,et al.BAFF/BAFF-R involved in antibodies production of rats with collagen-induced arthritis via PI3K-Akt-mTOR signaling and the regulation of paeoniflorin[J].JEthnopharmacol,2012,141(1):290-300.
[14] Chang Y,Zhang L,Wang C,et al.Paeoniflorin inhibits function of synoviocytes pretreated by rIL-1α and regulates EP4 receptor expression[J].JEthnopharmacol,2011,137(3):1275-82.
[15] Ranganathan P,McLeod H L.Methotrexate pharmacogenetics: the first step toward individualized therapy in rheumatoid arthritis[J].ArthritisRheum,2006,54(5):1366-77.
猜你喜欢
舰船科学技术(2022年10期)2022-06-17 06:26:42
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
建材发展导向(2021年14期)2021-08-23 00:57:20
临床医药文献杂志(电子版)(2017年11期)2017-05-17 04:48:47
当代医药论丛(2017年22期)2017-04-12 06:29:43
中国组织化学与细胞化学杂志(2016年3期)2016-02-27 11:15:41
中国卫生标准管理(2015年15期)2016-01-15 02:58:43
现代检验医学杂志(2015年6期)2015-02-06 01:44:07
西南医科大学学报(2014年6期)2014-03-20 15:43:47
当代畜禽养殖业(2014年11期)2014-02-27 08:00:01
