
量热分析化学放大方法及技术
2018-04-18 03:20:14郑艺华
分析测试学报 2018年3期
郑艺华
(青岛大学 机电工程学院 生物系统热科学研究室,山东 青岛 266071)
量热分析方法[1]通过测量各种热变化探索和建立内部反应现象、机理和规律,是进行反应热动力学研究,以及进行分析检测的主要方法和手段。比较而言,量热分析方法通用性强,不受光学及电化学等属性的干扰,满足实际试样的要求。
目前,各种类型的商用量热仪灵敏度和分辨率已达到微纳瓦级别,但价格昂贵、环境要求苛刻,主要在实验室进行理论分析和热力学、动力学研究。而在应用方面,以量热式生物传感器为代表的量热分析系统能准确而快速地进行分析测试[2-5],通过结合微流控分析和借助微纳米加工技术构建微纳米系统[6-7]是其重要的创新方向和潜在发展领域。
热信号是表观、宏观量,量热方法应用在痕量分析的特异性和灵敏度往往不尽如人意。一般地,其特异性可通过参比[8]并结合特定的生物敏感元件(酶、细胞等)来实现,但如果反应过程的热量变化很小,如α-糜蛋白酶催化ATEE水解的反应焓变仅为-1.1 kJ/mol,测量将非常困难,简单通过增大热传感器灵敏度[9]并结合预测算法[10]来提升灵敏度的方法有限,并不能提高信噪比。采用化学放大方法是一种有效途径,但仅从加大浓度等方面来考虑会增大系统比热,易造成沉淀,影响反应进程,通过富集减少样品量,降低检出限,进一步利用质子化反应和级联反应等手段增大反应热焓是最佳方案,灵敏度和选择性能的提高适用于分析检测定量化和小样化的发展趋势。
1 富 集
富集是分析预处理的必要步骤,是排除干扰、降低检出限、提高分析精度的重要手段,目前离线富集方式需手工操作,繁琐、耗时长,同时增加检测的复杂性和不确定性,也增加了误差传递过程。流动注射技术[11]的在线分离富集功能克服了手动操作的缺陷,使重现性和准确度显著提高。但仍需利用洗脱液,并且分离、富集和洗脱各步骤独立,流程和流路复杂,易干扰,效果受洗脱液性质制约。固相光度法自被Yoshimura等[12]提出以来,已广泛应用于各分析检测领域[13],该法集分离、富集、测试于一体,分析检测直接在固体基质表面进行,但由于采用可见光、紫外、荧光和光谱测试等光度方法,固相载体的透射或反射等光学性能限制了自身的选择范围及固相光度法的应用。

图1 富集、检测一体化量热式生物传感器系统原理图[17]Fig.1 Schematic diagram of the calorimetric biosensor with integrated of concentration and detection[17]1.six-way valve;2.sixteen-way valve;3.peristaltic pump;4.thermostat;5.injection valve;6.thermopile;7.reaction cell;8.reference cell;9.data acquisition module
通过相关基础微量热分析研究[14-15],集成富集的量热分析方法可同时实现复杂样品基体分离和目标组分富集,提高有效信号强度,并改善其特异性,满足对微量组分分析检测的需求。富集、检测一体化量热式生物传感器[16]省略了传统的洗脱等步骤,实现了在线分离、富集、反应、检测一体化,方法简单、快速、样品消耗量少,可实现自动化操作。如图1所示系统[17]用以测试重金属污染,反应室和参比室内填充富集载体,先富集样液的重金属,然后通入脲酶进行抑制,最后注入底物并在富集载体上发生酶反应,进行量热测定,有无重金属抑制条件下,脲酶活性发生变化,反映了重金属对脲酶反应的影响,进而得到重金属浓度,在0.002~0.01 mol/L浓度范围内,抑制率与铜离子的初始浓度接近线性关系,检出限为0.001 mol/L。
2 缓冲液放大
“缓冲液放大”是化学放大方法中最简单、最常用的方法,即利用缓冲溶液的质子化反应焓变实现热焓放大。假设存在反应(1),其过程中释放出n个质子。
X→Y+nH+;ΔHX,Y
(1)
当反应发生在缓冲液体系中,释放的质子会被对应的共轭碱捕捉并结合,同时伴随着质子焓变。
A-+H+→AH;ΔHr
(2)
那么反应过程总的焓变应为:
ΔHTotal=ΔHX,Y+ΔHr
(3)
所以,即使反应焓变很小,只要反应产生的质子被缓冲液捕获,发生并发质子化反应同时放出热量,反应焓变与质子化反应焓变共同作用仍会得到理想的反应焓变。如α-糜蛋白酶水解ATEE的反应在使用Tris缓冲液后,由于质子化放大作用,反应过程焓变由-1.1 kJ/mol增加为- 47.5 kJ/mol,热信号测量将非常容易。然而,质子化反应也可能带来问题,高质子化焓变缓冲液的使用可能产生相反的效应。例如,对于乳酸脱氢酶催化乳酸到丙酮酸,使用Tris缓冲液,测得的反应焓为-15.3 kJ/mol,如果使用磷酸盐缓冲液,测得的反应焓为- 47.3 kJ/mol[18]。故进行量热分析时,不一定选择高质子化焓变缓冲液,而需选择合适缓冲液种类。
基于反应设计和分析的需要,众多学者对质子化反应及焓变进行研究。在缺乏精密量热手段的时代,Bernhard等[19]通过文献值理论计算得到了Tris和磷酸盐缓冲液的质子化反应常数和热焓值;Roig等[20]量热分析了生物学研究常用的缓冲液(HEPES、PIPES、HEPPS、BES),并得到了不同温度状态下各缓冲液的质子化反应焓变;Jumean团队[21-25]对生化应用混合溶剂环境下的多种缓冲液(BES、TES、TABS、TAPS、TAPSO、MOPS、MOBS、MOPSO等) 的质子化反应及焓变进行了系列微量热研究。表1列出了20种常用缓冲液的质子化反应焓。

表1 不同缓冲液的质子化反应焓[26]Table 1 The protonation reaction enthalpy of different buffers[26]

图2 缓冲液体系的影响Fig.2 Influence of the buffer system
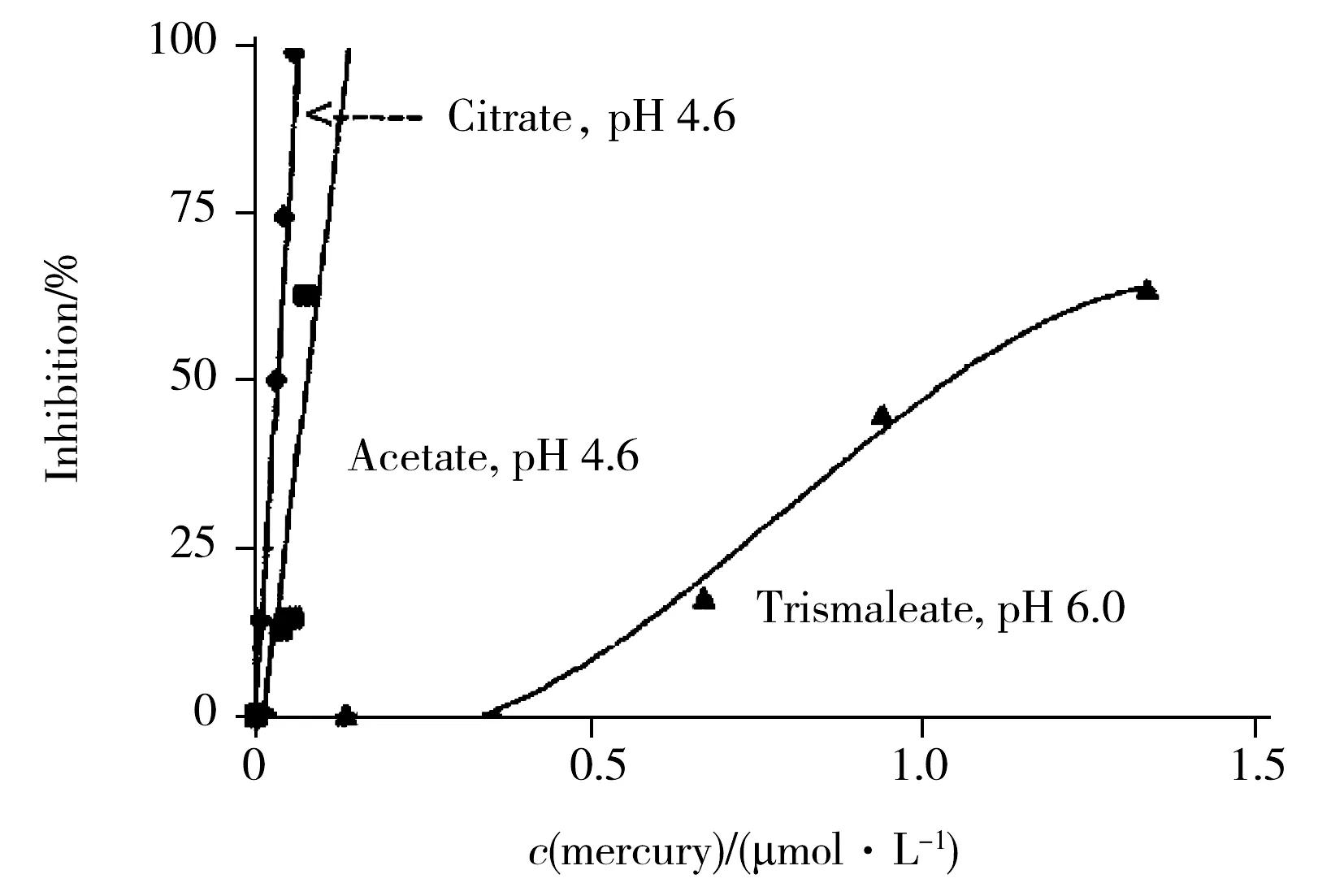
不同缓冲液在不同体系和不同催化反应过程的质子化反应和作用机理[27-31]可以应用到各量热分析领域。如胆碱酯酶抑制法是有机磷和氨基甲酸酯类农药残留检测的标准方法,但胆碱酯酶反应水解热较低,故普遍利用了磷酸盐缓冲液和Tris缓冲液进行热放大。Morgan等[32]使用磷酸盐缓冲液和固定化丁酰胆碱脂酶,利用酶热敏电阻实现了对水中有机磷和氨基甲酸酯农药的自动化连续监测,得到了不同农药种类与浓度和酶半衰期的时间关系;Mattiasson等[33]使用分流式酶热敏电阻,利用Tris缓冲液的质子化放大作用,分别采用农药水解酶和酶抑制方法对不同种类的有机磷农药进行了测定,检出限分别达10 mg/L和 1 mg/L;此外,脲酶抑制方法检测重金属具有优势,脲酶反应本身有理想的水解热值,通过缓冲液放大后可以达到60 kJ/mol[34]。如表2所示,不同缓冲液和pH值对脲酶水解反应焓变存在直接的影响,借助反应热变化得到的酶反应初速度来表示脲酶的活性,选择合适的缓冲液可以定量分析重金属离子 Cd2+、As3+、Zn2+,并可通过不同灵敏度实现特定重金属离子的鉴别[38-39],结果如图2所示。文献[40]也利用缓冲液放大研究了重金属离子的抑制作用,图3给出了citrate、acetate、tris-maleate 3种缓冲液下脲酶对重金属离子Cu2+、Hg2+抑制率的变化。由此可见,缓冲液热放大的研究结果对利用量热方法进行农药、重金属污染定量和半定量的快速分析具有较大的实用价值。
SolventCp(J·mL-1·K-1)RelativesensitivityAceticacid2 161 93Acetone1 702 46Benzene1 532 73Carbondisulphide1 622 58Carbontetrachloride1 363 07Chloroform1 442 901,4⁃Dichlorobenzene1 213 46Diethylether1 652 53Ethanol1 962 13Ethylacetate1 742 40n⁃Hexane1 492 80Methanol2 022 07Toluene1 562 68Water4 181 00
3 非水溶液体系
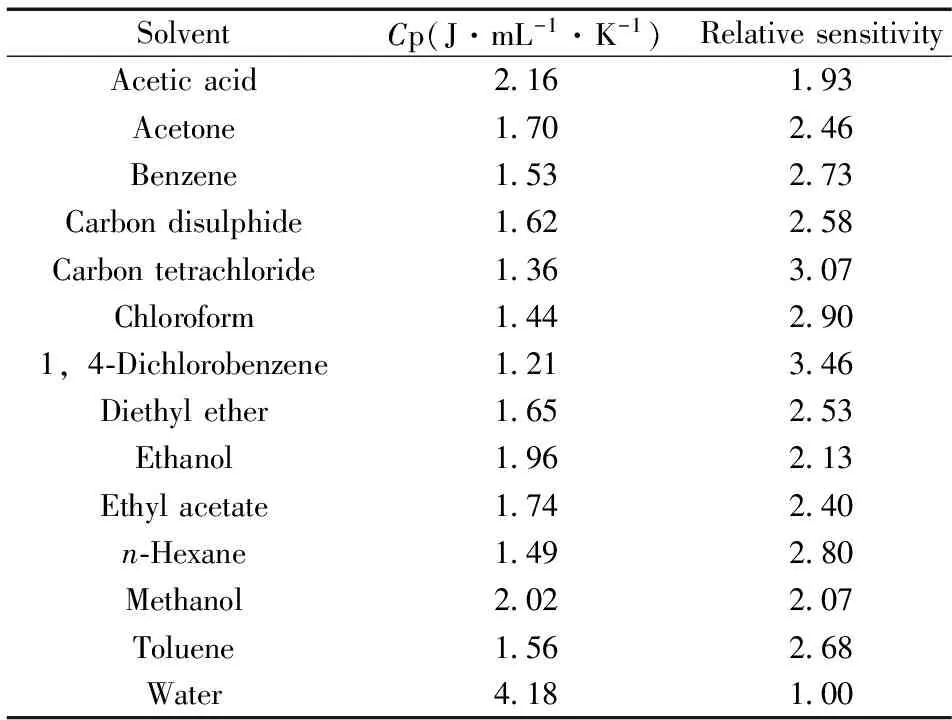
非水媒介反应在生物、化学和医学技术中的应用加强并扩展了对非水媒介反应特性的深入研究,目前量热分析[41-42]研究涉及各种有机溶剂组成的非水溶液体系(表3),并应用于胆固醇、过氧化物、甘油三脂和青霉素等的分析测量研究[43-47]。研究表明,由于较低的热容(数据见表3),非水溶液体系的量热灵敏度高于水溶液体系,缓冲液水溶液中存在适量的有机溶剂能使量热信号倍增。根据量热分析,通过溶剂氢键网格的影响,水的存在状态会极大地影响非水溶液体系以及缓冲液质子化反应的焓变[48-49]。
4 级联反应放大
级联反应放大是化学放大中最直接有效的方法,能提高有效信号强度,增大信噪比,降低检出限。早在1853年,Friedrich Mohr利用氯水氧化放大碘化物,能得到6倍的最初的量,后人采用次氯酸盐、溴水等氧化剂替代氯水深入研究了这种直接化学放大方法[50]。目前已出现组合酶放大[51]、聚合酶链式反应[52](PCR) 等技术,并广泛应用在检测、分析等领域。
利用酶的组合,可通过连续的酶反应来提高反应焓变,先前的产物可被连续地催化,最终得到的是焓变总和。如使用葡萄糖氧化酶(GOD)来测定葡萄糖[53-54],存在以下反应:
(4)
如果同时包含过氧化氢酶(Catalase,CAT),那么:
(5)
过氧化氢酶水解过氧化氢的热焓约为87 kJ/mol,反应总焓变有效提升,同时被葡萄糖消耗的氧可通过H2O2的分解部分再生,降低了H2O2的危害,并可增加线性范围。底物浓度较低时,尤其有效。级联反应(4)~(5)也用于纤维二糖水解酶水解纤维二糖的反应动力学热分析研究中,因纤维二糖水解酶水解纤维二糖的产物为葡萄糖,继续应用反应式(4)~(5),进行反应热焓放大,能够将其值从2.5 kJ/mol放大到360 kJ/mol,解决单独纤维二糖水解酶水解反应热量小,难以进行热分析研究的问题[55]。
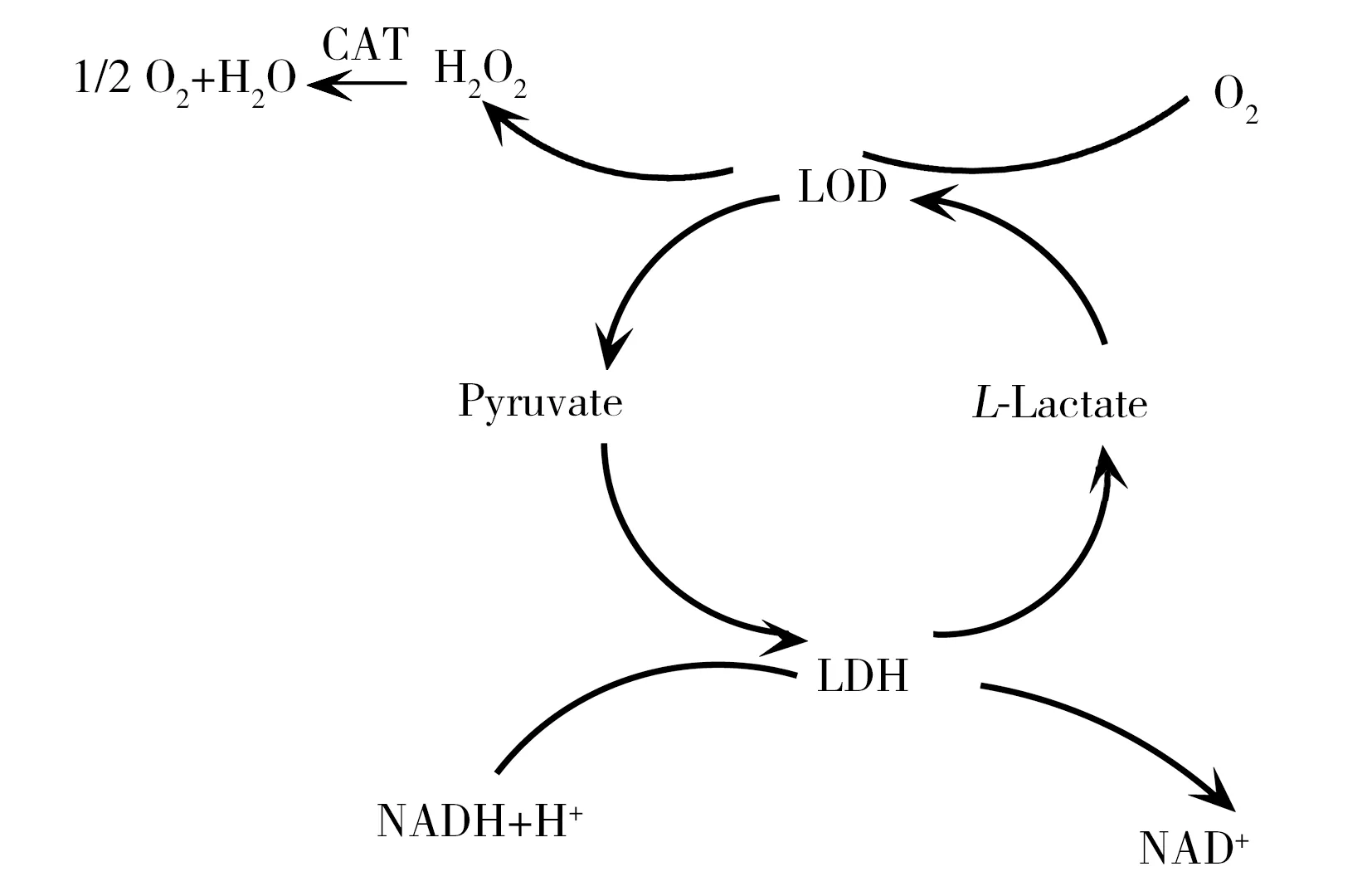
图4 乳酸氧化酶、乳酸脱氢酶和过氧化氢酶的放大系统[56]Fig.4 Amplification system composed by LDH,LOD and CAT[56]
另一种级联反应放大的方式是底物或副酶循环再生使用,通过预置副酶柱或同时固定多种酶来实现,可以提高灵敏度上千倍。Scheller等[56]采用此方法测定丙酮酸和乳酸,检出限达10 nmol/L,焓变高达-225 kJ/mol。它同时利用了乳酸脱氢酶(Lactic dehydrogenase,LDH)、乳酸氧化酶(Lactate oxidase,LOD)和过氧化氢酶,存在连锁反应(图4)。反应中乳酸被乳酸氧化酶氧化为丙酮酸(Pyruvate),然后丙酮酸被乳酸脱氢酶脱氢为乳酸(L-Lactate),周而复始,形成循环放大。同时,存在连续的过氧化氢酶的氧化反应,也放大了反应焓变。
农药残留和重金属污染等在环境中的存在浓度低,且状态和形式多样,若在现场对实际试样进行快速灵敏检测,除利用缓冲液对胆碱酯酶水解反应进行热放大外,采用酶级联反应也是有效手段。Bhand等[57]对热敏电阻装置进行了改进,增加了预置酶柱,其中放置乙酰胆碱酯酶(Acetylcholin esterase,AChE),AChE被农药抑制,产物胆碱的量减少并与被抑制的程度成正比,进一步地,胆碱的量可通过胆碱氧化酶(Choline oxidase,CHOD)和过氧化氢酶的级联反应热量得到,该检测利用了乙酰胆碱酯酶、胆碱氧化酶和过氧化氢酶的级联反应,其灵敏度较高,检测限为20~150 μg/L,其原理为反应式(6)~(8)。
(6)
(7)
(8)
Xu等[58]提供了相对廉价的农药残留量热分析解决方案,利用动植物体提取的替代酯酶和酚氧化酶完成级联酶反应,并通过离子交换法固定在树脂上,进行检测时,替代酯酶被农药抑制,然后利用酚氧化酶催化替代酯酶反应的产物酚类物质,反应热量变化增大了33倍,增强了热分析的灵敏度和性能。
Zheng等[59]将化学热放大原理应用于重金属检测,使用由醇氧化酶(Alcohol oxidase)和过氧化氢酶组成的固定化酶实现级联反应,醇氧化酶被重金属汞竞争性抑制,过氧化氢酶能催化醇氧化酶的水解产物——过氧化氢,通过级联反应,提升反应总热焓,大大提高检测灵敏度和分辨率。
5 总结与展望
化学放大方法和技术是突破量热分析方法选择性和灵敏度限制,提升其性能的根本有效手段和途径。量热分析方法可通过富集、缓冲液放大、非水溶液体系、级联反应等化学放大方法和手段从根本上提高量热分析的选择性、灵敏度和分析性能。上述热放大手段的使用可使一些测试的检测限从ppm级达到ppb级。
结合化学放大的量热分析方法能提供兼顾速度、灵敏度和准确性分析检测的有效方案和途径,尤其适合现场和快速痕量分析检测应用,以量热式生物传感器为代表的量热分析系统已应用于农药残留、重金属污染和安全分析监测等领域,借助于化学放大技术,量热分析方法将具备更广阔的应用前景。
参考文献:
[1] Peter J H.PrinciplesofThermalAnalysisandCalorimetry.Royal Society of Chemistry(Great Britain),Cambridge,2002.
[2] Bataillard P.TrACTrendsinAnal.Chem.,1993,12(10):387-394.
[3] Zheng Y H,Ma Y Z,Nie Z G,Yang Q R.JournalofSafetyandEnvironment(郑艺华,马永志,聂兆广,杨启容.安全与环境学报),2010,5(10):65-99.
[4] Zheng Y H,Hua T C,Sun D W,Xiao J J,Xu F.J.FoodEng.,2006,74:24-29.
[5] Yakovlevaa M,Bhandb S,Danielsson B.Anal.Chim.Acta,2013,766(5):1-12.
[6] Wang L,Sipe D M,Xu Y,Lin Q.JournalofMicroelectromechanicalSystems,2008,17(2):318-326.
[7] Lee W,Lee J,Koh J.NanobiosensorsinDiseaseDiagnosis,2012,1:17-29.
[8] Mattiasson B,Danielsson B,Mosbach K.Anal.Lett.,1976,9(10):867-889.
[9] Yoon J Y.Chapter4:TemperatureSensors,in:IntroductiontoBiosensors.New York:Springer,2013.
[10] Wang L Y,Zheng Y H.InstrumentTechniqueandSensor(王丽影,郑艺华.仪表技术与传感器),2013,2:1-3.
[11] Fang Z L.FlowInjectionSeparationandPreconcentration.New York:Wiley VCH,1993.
[12] Yoshimura K,Waki H,Ohashi S.Talanta,1976,23(6):449-454.
[13] Rocha F R P,Raimundo I M,Teixeira L S G.Anal.Lett.,2011,44(1/3):528-559.
[14] Wang L P,Xu Y,Zheng Y H,Li D X,Xu F.Chem.J.Chin.Univ.(王丽萍,胥义,郑艺华,李代禧,徐斐.高等学校化学学报),2012,33(8):1771-1776.
[15] Wang L P,Xu Y,Zheng Y H.Chem.J.Chin.Univ.(王丽萍,胥义,郑艺华.高等学校化学学报),2012,33(12):2638-2643.
[16] Zheng Y H,Liu J,Ma Y Z.Qingdao University.China Patent(郑艺华,刘君,马永志.青岛大学.中国专利),200910126677.9,2009-03-06.
[17] Zheng Y H,Liu X L,Ma Y Z,Xu Y,Xu F.Chin.J.Sci.Instrum.(郑艺华,刘晓林,马永志,胥义,徐斐.仪器仪表学报),2011,32(12):45-51.
[18] Rehak N N,Young D S.Clin.Chem.,1978,24:1414-1419.
[19] Bernhard S A.J.Biolog.Chem.,1956,218:961-969.
[20] Roig T,Baeckman P,Olofsson G.ActaChem.Scand.,1993,47:899-901.
[21] Bulos B N,Jumean F H.Thermochim.Acta,2004,411:91-94.
[22] Jumean F H,Abdo N M.Thermochim.Acta,2006,447:112-114.
[23] Jumean F H,Abdo N M.Thermochim.Acta,2006,448:44-46.
[24] Jumean F H,Abdo N M,Khamis M I.Thermochim.Acta,2008,475:22-24.
[25] Jumean F H,Abdo N M,Khamis M I.Thermochim.Acta,2011,524(1/2):194-196.
[26] Grime J K.AnalyticalSolutionCalorimetry.New York:Wiley-Interscience,1985.
[27] Sirotkin V A,Huettl R,Wolf G.Thermochim.Acta,2005,426:1-6.
[28] Bianconi M L.J.Biolog.Chem.,2003,278(21):18709-18713.
[29] Emilia O S,Carmen B,Luis G F.FEBSLetters,1998,435:219-224.
[30] Fukada H,Takahashi K.Proteins,1998,33:159-166.
[31] Wyrzykowski D,Pilarski B,Jacewicz D,Chmurzyn L.J.Therm.Anal.Calorim.,2013,111:1829-1836.
[32] Morgan W S G,Kuhn P C,Van N P P.WaterSci.Technol.,1985,17:855-865.
[33] Mattiasson B,Rieke E,Munnecke G.JournalofSolid-PhaseBiochemistry,1979,4(4):263-270.
[35] Hüttl R,Bohmhammel K,Wolf G,Oehmgen R.Thermochim.Acta,1995,250(1):1-12.
[36] Jespersen N D.J.Am.Chem.Soc.,1975,97(7):1662-1667.
[37] Schmidt H L,Krisam G,Grenner G.BiochimicaetBiophysicaActa(BBA)-Enzymology,1976,429(1):283-290.
[38] Oehlschläger K,Hüttl R,Wolf G.Thermochim.Acta,1996,271(10):41-48.
[39] Katrin O,Hüttl R,Wolf G.Thermochim.Acta,1998,310:185-189.
[40] Claudia P.Microchim.Acta,1999,130:209-214 .
[41] Flygare L,Danielsson B.Ann.NYAcad.Sci.,1988,542:485-496.
[42] Ramanathan K,Jonsson B R,Danielsson B.AnalysisinNon-aqueousMilieuUsingThermistors.In:Gupta,M.N.(Ed.),MethodsandToolsinBiosciencesandMedicine.MethodsinNon-AqueousEnzymology.BirkhauserVerlag,Basel,Switzerland,2000.
[43] Ramanathan K,Jonsson B R,Danielsson B.Anal.Chem.,2000,72:3443-3448.
[44] Danielsson B,Flygare L.Anal.Lett.,1989,22(6):1417-1428.
[45] Danielsson B,Flygare L.Sens.ActuatorsB:Chemical,1990,1(1/6):523-527.
[46] Hundeck H G,Wei M,Scheper T,Schubert F.Biosens.Bioelectron.,1993,8(3/4):205-208.
[47] Raghavan V,Ramanathan K,Sundaram P V,Danielsson B.Clin.Chim.Acta,1999,289:145-158.
[48] Goldberg R N,Kishore N,Lennon R M.J.Phys.Chem.Ref.Data,2001,30:1-13.
[49] Sirotkin V A,Hüttl R,Wolf G.J.Therm.Anal.Calorim.,2008,93(2):515-520.
[50] Rajagopalan S R.Bull.Mater.Sci.,1983,5(3/4):317-322.
[51] Hansen E H,Nørgaard L,Pedersen M.Talanta,1994,38(3):275-282.
[52] Kopp M U,Mello A J,Manz A.Science,1998,280(5366):1046-1048.
[53] Satoh I.Sens.ActuatorsB,1994,5(1/4):245-247.
[54] Salman S,Soundararajana S,Safina G,Satoh I,Danielsson B.Talanta,2008,77(2):490-493.
[55] Murphy L,Baumann M J,Borch K,Sweeney M,Westh P.Anal.Biochem.,2010,404:140-148.
[56] Scheller F,Siegbahn N ,Danielsson B.Anal.Chem.,1985,57:1740-1744.
[57] Bhand S,Werntoft K,Berg V,Högberg N,Sundaram P V,Danielsson B.ProceedingsofEighthWorldCongressonBiosensors,Granada Spain,2004.
[58] Xu F,Jiang M,Xu Y,Hua Z Z,Ma T H.Shanghai University of Science and Technology.ChinaPatent(徐斐,姜觅,胥义,华泽钊,马天画.上海理工大学.中国专利),200610147245.2,2006-12-14.
[59] Zheng Y H,Liu J,Ma Y Z.Qingdao University.China Patent(郑艺华,刘君,马永志.青岛大学.中国专利),201110150133.3,2011-05-25.
猜你喜欢
昆明医科大学学报(2020年12期)2021-01-26 00:44:12
电子制作(2016年15期)2017-01-15 13:39:09
西安文理学院学报(自然科学版)(2016年4期)2016-12-19 08:18:59
China International Studies(2016年3期)2016-07-14 03:00:06
系统工程与电子技术(2016年2期)2016-04-16 05:16:51
中国粮油学报(2016年5期)2016-01-23 02:44:40
质谱学报(2015年5期)2015-03-01 03:18:25
电测与仪表(2014年1期)2014-04-04 12:00:34
电测与仪表(2014年1期)2014-04-04 12:00:28
茶叶通讯(2014年2期)2014-02-27 07:55:39
