
通过式固相萃取净化/UPLC-MS/MS法测定特殊医学用途配方食品中的13种非法添加化学成分
2018-04-18 03:22:14邱红钰贾彦博王红青
分析测试学报 2018年3期
励 炯,邱红钰,贾彦博,王红青,李 玮
(杭州市食品药品检验研究院,浙江 杭州 310017)
糖尿病是一种以胰岛素分泌或作用缺陷引起的高血糖为特征的代谢性疾病,易引起患者的糖、蛋白质、脂肪、水和电解质等一系列代谢紊乱[1]。预计到2030 年,中国的糖尿病患病率将达11.6%,居各国之首,且以老年人为主,呈日益增多的趋势[2]。目前,针对糖尿病患者的保健食品和特殊医学用途配方食品(Food for special medical purpose,FSMP)种类繁多,常应用于糖尿病、呼吸系统综合疾病、肾病、肿瘤、炎性肠病、食物蛋白过敏、肥胖、先天性代谢缺陷等病症的营养辅助[3-4]。虽然近几年国家卫计委出台了《GB 29922-2013 特殊医学用途配方食品通则》和《GB 29923-2013 特殊医学用途配方食品良好生产规范》,国家食品药品监督管理总局也于2016年7月1日实施颁布《特殊医学用途配方食品注册管理办法》,进一步规范和管理FSMP的注册,但相应的监督管理和法律责任等不完善。近年来已有很多文献对中成药和保健食品中非法添加的降糖类化学成分进行研究[5-7],发现一些不法商家在利益驱动下,为了增强降糖效果,可能会在针对糖尿病人的FSMP产品中非法添加各种降糖类化学药物,患者往往在不知情的情况下长期服用,造成不可挽救的后果。
目前,检测化学降糖成分的方法主要有薄层色谱法[8-9]、高效液相色谱法[10-12]、毛细管电泳法[13]、离子选择性电极法[14]、气相色谱-质谱法[15]、液相色谱-质谱联用法[16-18],其中液相色谱-质谱联用法兼具灵敏度高及抗干扰性强的优势,广泛应用于降糖类非法添加化学成分的定性确证。
样品提取和净化作为关键技术,会直接影响方法灵敏度和精密度,尤其是部分FSMP配料中脂肪或者磷脂含量较多,基质效应较明显。前处理净化方法多以普通的固相萃取小柱净化,但其步骤繁琐,操作不当易造成损失而影响定量准确性。本研究采用通过式固相萃取技术,以Oasis PRiME HLB 固相萃取柱净化样品,上样前无需活化和平衡,样液直接加载至SPE柱以吸附脂肪和磷脂类化合物,建立了针对糖尿病患者的特殊医学用途配方食品中13种非法添加降糖类化学成分的通过式固相萃取/超高效液相色谱-串联质谱检测方法,方法灵敏、简便、高效,可满足日常特殊医学用途配方食品的质量监测需求。
1 实验部分
1.1 仪器与试剂
美国Agilent 1290高效液相色谱仪、Agilent 6495串联四极杆质谱仪、Milli-Q去离子水发生器(美国Millipore公司)、MS3旋涡混合器(德国IKA公司)。
对照品甲苯磺丁脲(批号90126,质量分数99.0%)购于德国Dr.Ehrenstorfer GmbH;格列本脲(批号10135-200404,质量分数100%)、格列齐特(批号100269-201004,质量分数99.9%)、格列吡嗪(批号100281-200602,质量分数99.4%)、格列喹酮(批号100280-201002,质量分数99.3%)、格列美脲(批号100674-200301,质量分数99.1%)、马来酸罗格列酮(批号100952-200701,质量分数99.5%)、瑞格列奈(批号100753-200501,质量分数99.8%)、盐酸吡格列酮(批号100634-201202,质量分数100%)、盐酸二甲双胍(批号100664- 201203,质量分数100%)、盐酸苯乙双胍(批号100922-201001,质量分数99.7%)、格列波脲(批号520026-201401,质量分数99.9%)购于中国食品药品检定研究院;盐酸丁二胍(批号1-RLJ-70-2,质量分数96%)购于Toronto Research Chemicals Inc.。冰乙酸、乙酸铵(分析纯)购自国药集团化学试剂有限公司;甲醇、乙腈、甲酸(色谱纯,德国Merck公司);Oasis PRiME HLB 固相萃取柱( 6 mL中含200 mg,美国Waters公司)。
1.2 标准溶液的配制
分别精密称取13种降糖对照品标准品各10 mg,分别用甲醇溶解,转移至10 mL容量瓶中,用甲醇定容,作为对照品储备溶液(质量浓度为1 000 mg/L)。分别精密吸取上述对照品储备溶液适当体积于10 mL容量瓶中,用甲醇稀释定容,配成2.0、5.0、10、20、50、100 μg/L的混合标准品系列溶液。
1.3 样品前处理
取2 g样品置50 mL聚氟乙烯离心管中,精密加入10 mL含1%甲酸的90%乙腈水溶液,加入2 g氯化钠和5 g无水硫酸钠,涡旋混合1 min,超声提取20 min,5 000 r /min 离心5 min。取8 mL提取液直接加载到PRiME HLB 固相萃取柱上,流速为1滴/s,收集全部流出液。准确移取5.00 mL流出液,40 ℃水浴下氮吹至近干,精密加入0.5 mL 5 mmol/L乙酸铵水溶液-甲醇(80∶20,体积比),涡旋混匀1 min溶解残渣,用0.22 μm微孔滤膜过滤。
1.4 色谱与质谱条件
色谱柱:Waters ACQUITY UPLC BEH C18(150 mm×2.1 mm,1.7 μm);流动相:A为含0.1%甲酸的5 mmol/L 乙酸铵溶液,B为甲醇;梯度洗脱程序:0~10 min,80%~20% A;10 ~10.1 min,20%~80% A ;10.1~13 min,80% A。柱温:40 ℃;流速:0.4 mL/min,进样量1 μL;运行时间:13 min。
离子源:电喷雾离子源(ESI),正离子检测方式(ESI+);多反应检测(MRM模式);干燥气温度:200 ℃;干燥气流速:14 L/min;毛细管电压:3.5 kV;毛细管出口电压(Fragmentor):380 V;校准方法:质量轴自动调谐校正;MRM进行分段扫描:0~1.4 min,盐酸二甲双胍;1.4~2.4 min,盐酸丁二胍;2.4~4.5 min,盐酸苯乙双胍;4.5~6.0 min,甲苯磺丁脲;6.0~7.0 min,格列吡嗪和格列齐特;7.0~8.8 min,马来酸罗格列酮、格列波脲、吡格列酮、格列本脲和格列美脲;8.8~9.4 min,格列喹酮;9.4~13.0 min,瑞格列奈。其他质谱分析参数见表1。
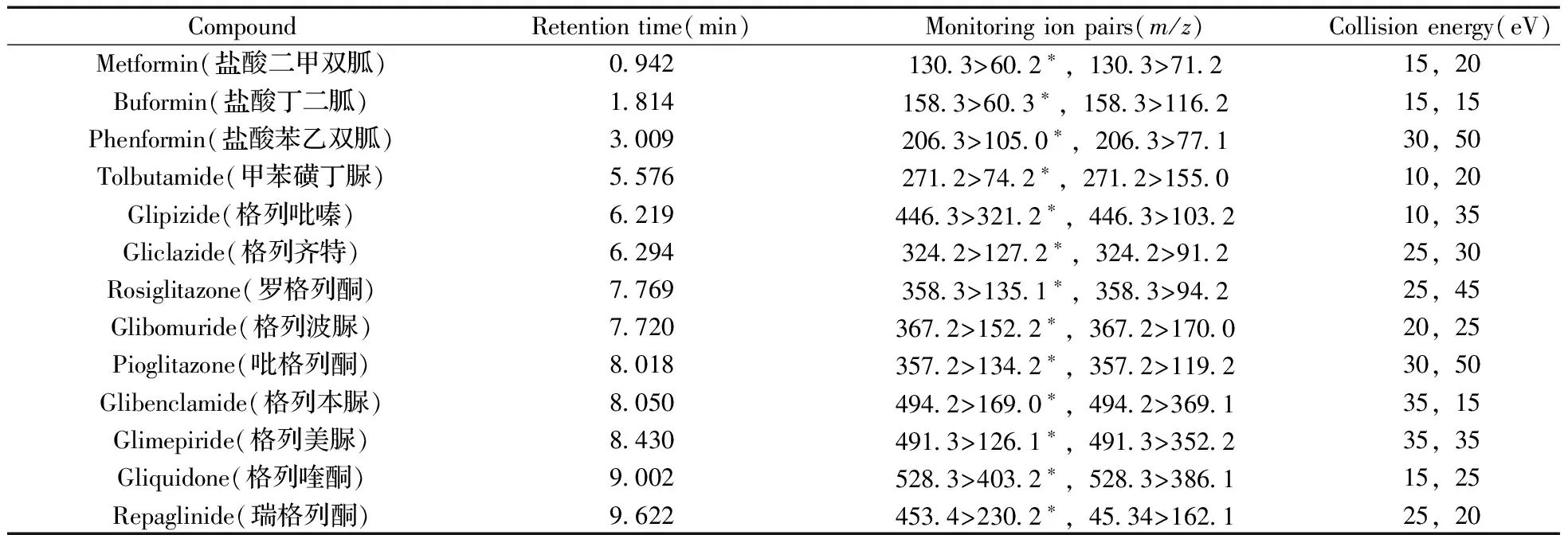
表1 13种降糖类化学成分的质谱分析参数Table 1 MS parameters of 13 anti-diabetic drugs
*quantitative ion pair
2 结果与讨论
2.1 质谱条件的优化
精密量取质量浓度为0.5 mg/L的13种目标化合物标准溶液分别注入进样器,随流动相直接注入质谱仪进行母离子和子离子扫描。研究发现,13种降糖类化学成分在正离子模式下的方法稳定性和重复性较负离子模式好,故本方法采用正离子模式进行检测。
在正离子模式下对13种降糖类化学成分进行母离子扫描,扫描范围为m/z100~450,得到 [M+H]+峰。确定母离子之后,调节毛细管电压,当毛细管电压为3.5 kV时一级扫描响应最高。确定母离子及其条件后,继续进行子离子扫描,以得到最佳的二级质谱条件,经仪器自带的二级质谱参数自动优化程序,得到13种目标化合物的二级质谱优化参数(表1)。最终将优化后得到的定量定性离子对和碰撞能量作为13种降糖类化学成分的扫描参数。
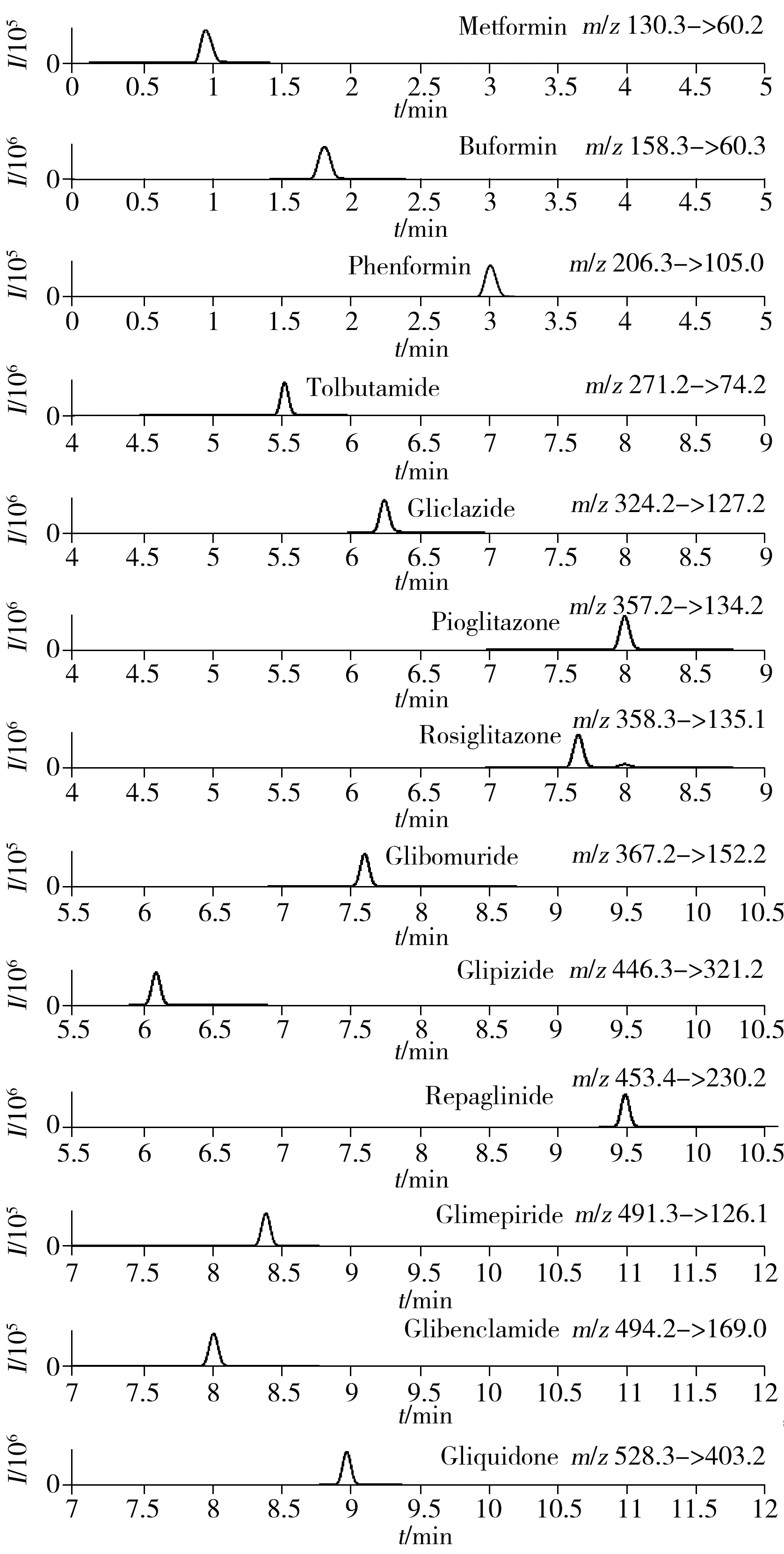
图1 13种降糖类化学成分(10 μg/L)的MRM谱图Fig.1 MRM chromatograms of 13 anti-diabetic drugs(10 μg/L)
2.2 色谱条件的优化
本文比较了Waters ACQUITY UPLC BEH C18(150 mm×2.1 mm,1.7 μm)、Waters CORTECS C18(150 mm×2.1 mm,1.6 μm)、Agilent ZORBAX Eclipse Plus C18(100 mm×2.1 mm,1.8 μm)和Kinetex C18(100 mm×2.1 mm,1.7 μm ) 4种色谱柱对13种降糖类化学成分色谱行为的影响。盐酸二甲双胍、盐酸苯乙双胍和盐酸丁二胍这3种双胍类降糖成分,均为含氮类化合物,在色谱柱上易产生二次保留从而使色谱峰拖尾。结果表明,使用Waters ACQUITY UPLC BEH C18色谱柱时13种降糖类化学成分的色谱峰峰形较好,而采用其他3种色谱柱分析时,3种双胍类成分均产生比较严重的色谱峰拖尾现象。这是因为Waters ACQUITY UPLC BEH色谱柱采用亚乙基桥杂化颗粒技术,可以保证目标物较好的峰对称性、柱效以及化学稳定性,适用于最广泛的化合物种类的通用性,在一定程度上解决了在质谱上较难分析双胍类降糖成分的问题。
本文考察了甲醇-0.1%甲酸水溶液、甲醇-5 mmol/L乙酸铵溶液以及甲醇-含0.1%甲酸的5 mmol/L乙酸铵溶液3组流动相在“1.4”梯度条件下的色谱行为。实验结果显示:甲醇-0.1%甲酸水溶液的目标峰峰形较好,但灵敏度偏低;甲醇-5 mmol/L乙酸铵流动相的目标峰灵敏度明显提高,但峰形拖尾。本文采用甲醇-含0.1%甲酸的5 mmol/L乙酸铵溶液作为流动相,此时13种目标物的峰形较好,灵敏度较高,MRM谱图见图1。
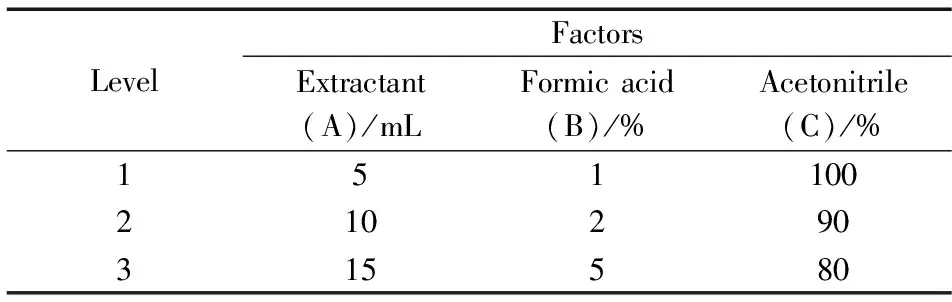
表2 正交试验因素水平Table 2 Orthogonal test factors and levels
2.3 提取及净化方式的优化
降糖类化学成分的提取溶剂一般采用甲醇、乙腈等,而一般通过式固相萃取法提取药物残留多采用乙腈为提取溶剂,在提取剂中加入一定比例的甲酸,有助于沉淀样品中的蛋白质。本研究通过对提取剂用量、甲酸比例以及乙腈浓度(分别作为因素A、B、C)进行正交试验优化(每个因素取3个水平,按照L9(33)正交表试验),比较10 μg/L加标水平下各因素组合的回收率和净化效果,选择最佳配比组合。正交试验因素见表2。结果表明,使用10 mL含1%甲酸的90%乙腈水溶液为提取剂可以获得良好的提取和净化效果。
除了蛋白质,脂类物质也是主要的基质干扰物,本文采用通过式固相萃取技术,提取液经Oasis PRiME HLB 固相萃取柱净化,上样前无需活化和平衡,样液直接加载至SPE柱以吸附脂肪和磷脂类化合物,不仅有效净化了样品溶液,而且显著提高了样品分析通量。
2.4 基质效应评估
本文通过基质效应来评价采用的通过式固相萃取技术的净化效率。基质效应(ME)以基质标准曲线的斜率与溶剂标准曲线的斜率之比进行评估,基质标准曲线配制如下:分别取6份空白样品2.0 g(检测结果为阴性的降糖奶粉)于50 mL聚丙烯离心管中,分别精密加入混合对照品储备溶液(质量浓度为1 mg/L)适量,按“1.3”方法制备基质标准曲线,配成最终质量浓度为2.0、5.0、10、20、50、100 μg/L的基质标准品系列溶液,按“1.4”条件进行分析,得到以降糖奶粉为基质的基质标准曲线,所得的基质标准曲线斜率与溶剂标准曲线斜率的百分比进行评估。一般情况下,ME在85%~115%之间不存在明显的基质效应,本文结果ME为88.4%~96.7%,所以本方法较好地消除了基质效应,保证了定性、定量结果的准确可靠。
2.5 线性关系、定量下限与重复性
取“1.3”方法配制的标准品系列溶液,分别进样1 μL,以定量监测离子对的离子流中谱峰峰面积(Y)为纵坐标,标准品质量浓度(X,μg/L)为横坐标进行线性关系考察,结果见表3。13种降糖类化学成分在2~100 μg/L范围内线性较好(r2均大于0.99)。
取空白的降糖类奶粉样品2.0 g,加入适当稀释的混合标准品溶液,按“1.4”方法制备供试品溶液,分别以信噪比为3和10计算13种降糖类化学成分的检出限(LOD)与定量下限(LOQ),结果见表3。13种降糖类化学成分的最低检出限为0.10~0.35 μg/kg,定量下限为0.3~1.0 μg/kg。
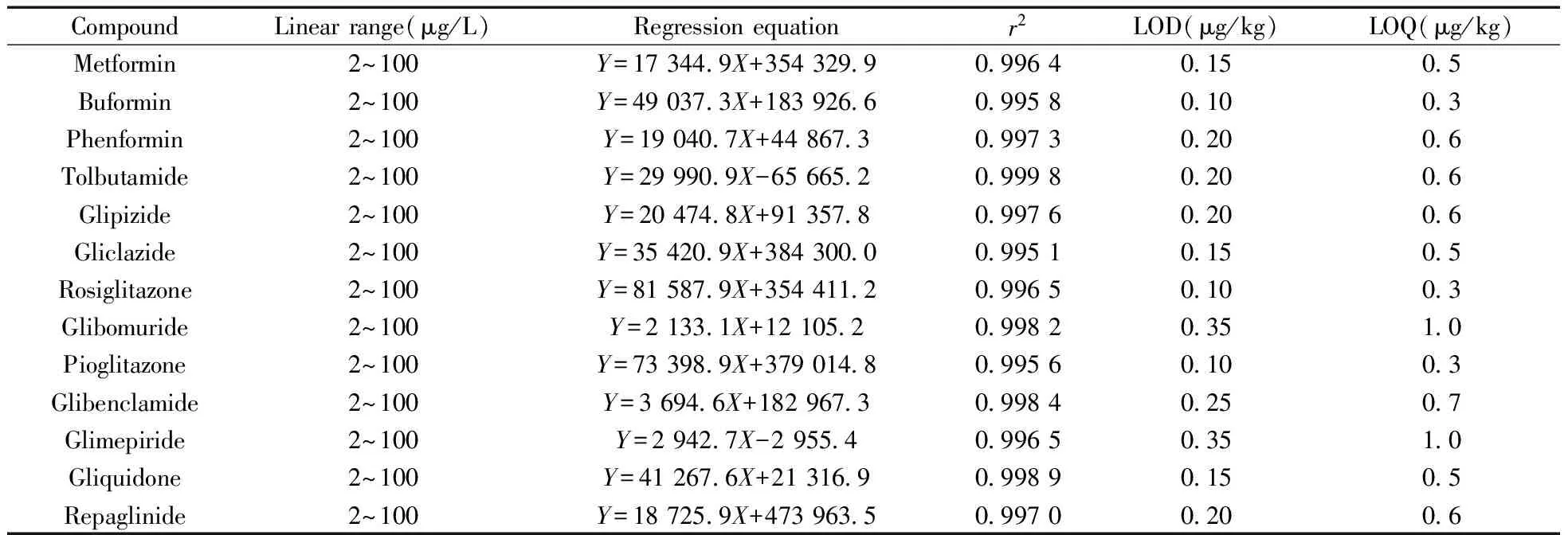
表3 13种降糖类化学成分的线性关系、检出限和定量下限Table 3 Linear regression results,LODs and LOQs of 13 anti-diabetic drugs
2.6 方法回收率与精密度
取空白降糖奶粉样品,每份称取2.0 g,分别加入适量的混合对照品溶液,使供试品溶液最终加标水平分别为2、5、20 μg/kg,平行6份,按样品制备方法制备供试品溶液,检测含量,回收率结果见表4。13种降糖类化学成分的平均回收率为80.9%~102.8%,相对标准偏差(RSD)为2.6%~8.2%。
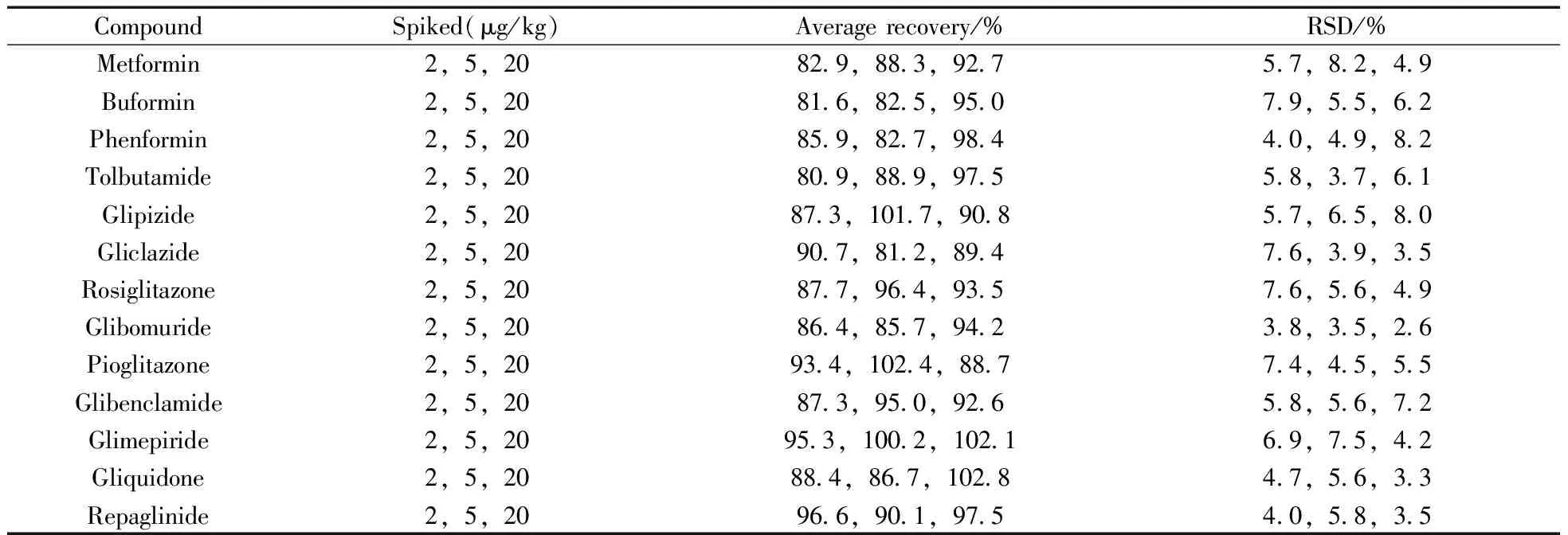
表4 特殊医学用途配方食品中13种降糖类化学成分的回收率与精密度(n=6)Table 4 Recoveries and precisions of 13 anti-diabetic drugs in FSMP(n=6)
2.7 实际样品检测
利用本文建立的方法检测了5批次降糖奶粉(分别为雅培益力佳SR营养配方粉、雅培怡保康营养配方粉、雀巢SUSTAGEN低GI营养配方奶粉、营养狮牌糖友宝配方奶粉和阿尔发牌富铬奶粉)、2批次降糖饼干(分别为阿尔发牌降糖饼干和思朗牌无添糖消化饼干)和1批次降糖麦片(阿尔发牌消渴麦片),结果显示,13种降糖类化学成分均未检出。
3 结 论
本研究建立了通过式固相萃取净化/超高效液相色谱-串联质谱法测定特殊医学用途配方食品中13种非法添加降糖类化学成分的分析方法,优化了色谱质谱条件以及提取净化条件。方法无需复杂的样品前处理,样品经含1%甲酸的90%乙腈水溶液提取,使用Oasis PRiME HLB固相萃取柱进行净化处理,有效降低了基质效应,提高了分析方法的选择性和灵敏度。
参考文献:
[1] Hu Y Y,Huang L H.ChineseJournalofNursing(胡一宇,黄丽华.中华护理杂志),2013,48(6):555-557.
[2] Peng Y M,Qin Y Q,Gong N C,Li Y H.J.FoodSci.Technol.(彭艳梅,覃元清,龚年春,李跃辉.食品科学技术学报),2017,35(1):17-20.
[3] Suo S Z,Hu H,Wang Y T.Chin.J.FoodHyg.(索思卓,胡豪,王一涛.中国食品卫生杂志),2016,28(2):182-186.
[4] Chen B,Dong H S,Zhang G W,Qin Y Q,Zang P,Yu Y B,Zhang S J.J.FoodSci.Technol.(陈斌,董海胜,张国文,覃元清,臧鹏,于燕波,张淑静.食品科学技术学报),2017,35(1):6-16.
[5] Sui H S,Wang L J,Xie F.Chin.Pharm.(隋海山,王立娟,谢菲.中国药业),2015,(22):110-112.
[6] Wang W,Mao Q.GuangzhouChem.Ind.(王巍,毛庆.广州化工),2016,43(22):120-121.
[7] Ning X,Zhang W Q,Wang G L,Cao J,Zhang Q S.J.FoodSaf.Qual.(宁霄,张伟清,王钢力,曹进,张庆生.食品安全质量检测学报),2015,(5):1876-1882.
[8] Shweta S H,Sunil R D.J.Liq.Chromatogr.Relat.Technol.,2011,34(2):966-980.
[9] Wang Q,Su J,Qin H P,Lu J.Chin.Pharm.Affairs(汪祺,苏晶,覃红萍,鲁静.中国药事),2010,24(7):647-649.
[10] Li J,Cao Q W,Wang J F,Hu M J,Jiang H,Jia Y B.ChineseTraditionalandHerbalDrugs(励炯,曹青文,王姣斐,扈明洁,江海,贾彦博.中草药),2017,48(13):2666-2673.
[11] Zhang X D,Chen J,Wu X Y,Gu Z L,Zhou G H.Chin.J.Pharm.Anal.(张晓丹,陈静,伍小勇,古卓良,周国华.药物分析杂志),2011,(6):1088-1093.
[12] Wang D,Yang X,Cheng Y F,Su K,Yang B Q,Liu H J.Chin.J.Pharma.Anal.(王豆,杨欣,程怡凡,苏珂,杨斌齐,刘海静.药物分析杂志),2013,33(8):1377-1381.
[13] Znaleziona J,Maier V,Ranc V,Sevcik J.J.Sep.Sci.,2011,34(10):1167-1173.
[14] Waheed A B,Mohammed A E,Inas M M.Anal.Sci.,2009,25(12):1431-1436.
[15] Wang J,Yang Z M,Kong H W,Li Y,Lu X,Xu G W.Chin.J.Chromatogr.(王静,袁子民,孔宏伟,李勇,路鑫,许国旺.色谱),2012,30(1):8-13.
[16] Wu X M,Zhu B H,Lin L,Huang W X,Pang D W.FoodChem.,2012,133(2):482-488.
[17] Zhu F,Ruan L P,Ma Y J,Ji W L,Liu H L.Chin.J.Chromatogr.(朱峰,阮丽萍,马永建,吉文亮,刘华良.色谱),2014,32(1):13-20.
[18] Du Y S,Li Q,Wu C M,Zhang Y.Chin.J.Chromatogr.(杜彦山,李强,吴春敏,张岩.色谱),2015,33(4):371-376.
猜你喜欢
基层中医药(2020年6期)2020-09-11 06:35:28
中成药(2018年12期)2018-12-29 12:25:38
中成药(2018年6期)2018-07-11 03:01:14
中国蜂业(2018年4期)2018-05-09 06:25:08
基层中医药(2018年1期)2018-03-01 07:36:13
天然产物研究与开发(2016年1期)2016-06-05 10:29:22
中国民族医药杂志(2016年7期)2016-05-09 07:49:19
当代化工研究(2016年6期)2016-03-20 16:21:46
上海农业学报(2016年5期)2016-02-10 06:53:10
无机化学学报(2014年3期)2014-02-28 17:30:58
