
U-PLS/RBL三维荧光法测定藤络宁胶囊中东莨菪内酯含量
2018-04-18 03:20:09刘德龙魏永巨
分析测试学报 2018年3期
刘 铁,刘德龙,魏永巨
(河北师范大学 化学与材料科学学院,河北 石家庄 050024)
藤络宁胶囊是由丁公藤、羌活、独活、防己、延胡索、丹参等组成的复方制剂,具有疏风散寒、除湿通络的功用,用于类风湿性关节炎属寒湿瘀阻证[1]。而东莨菪内酯作为处方中君药丁公藤的主要有效成分之一,具有镇痛及抗炎等作用[2-7],故测定其含量具有实际意义。目前,测定藤络宁胶囊中东莨菪内酯含量的方法只有高效液相色谱法(HPLC)[8]。国标[1]中采用高效液相色谱梯度洗脱,分析过程耗时较长。
实验研究表明,东莨菪内酯有强荧光性质,而荧光方法具有灵敏度高、测试简单快速的优点,但是若测试组分与共存组分的光谱重叠时选择性较差,化学计量学中二阶校正方法的“二阶优势”可以弥补该缺点进行定量分析。解析三维荧光矩阵有效的二阶校正方法主要包括平行因子分析法(PARAFAC)、三线性分解(ATLD)及其衍生方法(交替惩罚三线性分解法APTLD,自加权交替三线性分解法SWATLD),以及基于潜变量的残差双线性分解的偏最小二乘法(包括Unfolded partial least squares coupled to residual bilinearization(U-PLS/RBL)和Multi-way partial least squares coupled to residual bilinearization(N-PLS/RBL))[9]。前两类解析方法既能获得分析体系的定性信息(激发光谱和发射光谱),又能进行定量分析[10-12],而基于潜变量的偏最小二乘法则只能获得定量分析结果。但有一些文献报道发现,U-PLS/RBL和N-PLS/RBL在解析组分之间或者组分与背景之间严重重叠及存在内滤光效应等的复杂光谱体系时,能够给出更好的定量分析结果[13-19]。目前尚未见利用二阶校正方法U-PLS/RBL解析三维荧光测定藤络宁胶囊中东莨菪内酯含量的报道。
本文首先研究了东莨菪内酯的荧光性质及最佳实验条件,选择最佳测定波长范围后,用U-PLS/RBL解析合成样确定了方法的可靠性,然后用于实际样品藤络宁胶囊中东莨菪内酯的定量分析。为了验证方法的可靠性,用HPLC方法进行了确证。
1 U-PLS/RBL算法原理
首先测定一系列校正样和预测样的三维荧光光谱矩阵,然后按照如下步骤进行计算。
第一步校正:即在校正样的浓度和光谱数据之间建立PLS模型。先将测定的每个校正样的三维荧光矩阵向量化(图1左半部分),即将每个三维荧光矩阵Xi(J,K)转化为向量xi(J×K,1),其中i=1,2,…,I,I为校正样个数,J和K分别为激发和发射波长个数,之后将所有向量排成矩阵形式X(J×K,I)。然后用X(J×K,I)与校正样中的浓度向量y(I× 1)进行PLS建模,得到两个载荷矩阵P(JK×A)、W(JK×A)及回归系数向量v(A× 1),A为利用基于去一法的交叉验证法得到的潜因子数[20]。v用于估计待测样中相应组分的浓度。
第二步预测:如果待测样中不含其它未知成分,则按照如下公式准确估计待测样中相应组分的浓度。
(1)
tu=(WTP)-1WTvec(Xu)
(2)
其中,tu为待测样向潜变量空间的投影得分向量,vec(Xu)为待测样矩阵的向量化形式。
如果待测样中含有其它未知成分,由式(2)计算的tu不适合计算待测样中的组分浓度,表现为PLS预测残差远大于仪器噪声。此时按照基于主成分分析的残差双线性分解法(RBL)对背景信号进行扣除[9],公式为(图1右半部分):
Xu=reshape(PtRBL)+BRBLTRBLT+ERBL
(3)
上式说明样本数据由两部分构成,一部分为校正样组分的信号,另一部分则来自试样中某些潜在的干扰组分。reshape表示将向量转化为矩阵,ERBL表示误差矩阵,BRBLTRBLT表示对残差矩阵(Xu-reshape(PtRBL))进行主成分分析后的矩阵乘积(主成分数=Nunx)。
对式(3)中ERBL的模进行极小化(P保持不变,只改变tRBL),得到最佳的tRBL,将tRBL代入式(1)可得到扣除背景信号后的真实浓度。详细的原理和算法见文献[9]。U-PLS/RBL解析过程如图1所示[9]。
2 实验部分
2.1 仪器与试剂
F-4600型荧光分光光度计(日本日立公司,用1 cm石英比色皿扫描激发-发射三维荧光光谱);激发波长:310~420 nm,发射波长:400~550 nm。狭缝宽度为5.0/5.0 nm,扫描速度为1 200 nm/min。
安捷伦1260高效液相色谱仪(配FLD检测器);分析天平(0.000 01 g);pH酸度计(美国Orion公司,Orion868)。
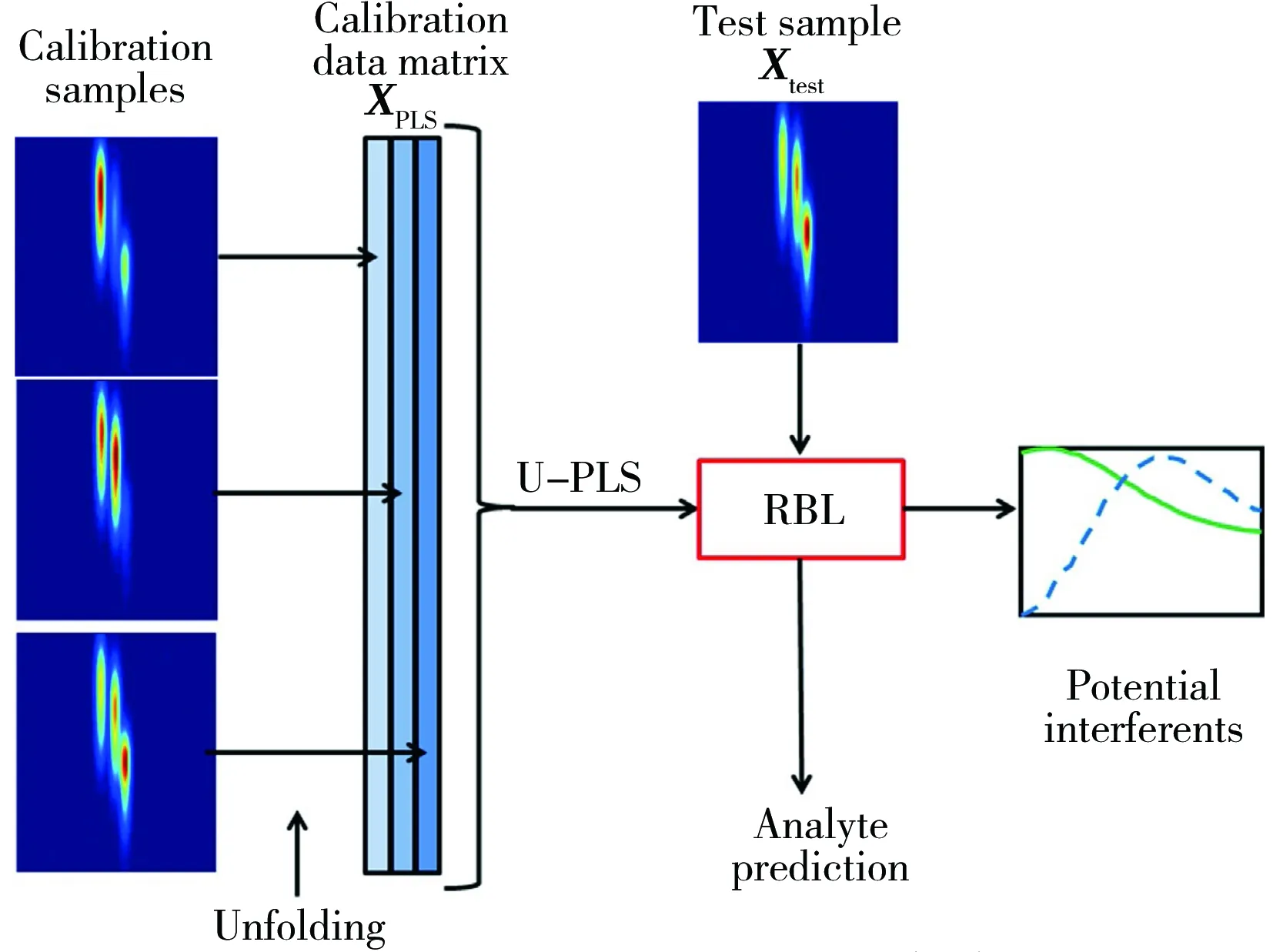
图1 U-PLS/RBL解析过程Fig.1 Scheme illustrating how U-PLS/RBL works for second-order data three typical calibration samples(of a possibly much larger calibration set) are shown at the left,from which the unfolding operation leads to the XPLS calibration data matrix;the latter is processed with U-PLS and submitted,together with the test sample data X test,to RBL for separating the contribution of the potential interferents,yielding accurate analyte prediction
东莨菪内酯(上海源叶生物科技有限公司):精确称取1.39 mg东莨菪内酯标准品,用甲醇溶解并定容于25 mL容量瓶中,配成55.6 mg/L的操作液,在4 ℃冰箱避光保存。
藤络宁胶囊(购于上海天猫国大药房旗舰店,生产厂家:吉林省通化博祥药业股份有限公司):取出藤络宁胶囊内的药粉,准确称取0.980 0 g。甲醇浸泡超声1 h后,摇匀、过滤,并将滤液转至50 mL棕色容量瓶,用甲醇定容,得到19.6 g/L的操作液,置于冰箱4 ℃避光保存。
甲醇(色谱纯);磷酸(分析纯);BR缓冲溶液(0.4 mol/L,pH 9.0);实验所用其他试剂均为分析纯,实验用水为自制二次蒸馏水。
2.2 实验方法
2.2.1合成样的测定取一系列10 mL容量瓶,加入2.00 mL BR缓冲溶液、东莨菪内酯溶液和一定量的甲醇,用二次蒸馏水定容后混合均匀。其中7个校正集溶液东莨菪内酯质量浓度为0.014 7、0.029 4、0.044 1、0.058 8、0.073 5、0.088 2、0.102 9 mg/L;配制6个确证样,其中东莨菪内酯质量浓度为0.022 05、0.036 75、0.051 45、0.066 15、0.080 85、0.095 55 mg/L。
2.2.2藤络宁胶囊中东莨菪内酯含量的测定取一系列10 mL容量瓶,加入2.00 mL BR缓冲溶液、东莨菪内酯以及适量藤络宁胶囊提取液,用二次蒸馏水定容后混合均匀,进行三维荧光扫描。
其中1~7号为校正集,校正样中东莨菪内酯质量浓度分别为0.014 7、0.029 4、0.044 1、0.058 8、0.073 5、0.088 2、0.102 9 mg/L。8~12号为样品集,其中含有一定浓度的藤络宁胶囊提取液。
2.2.3回收率实验1~7号为校正集,浓度同上。8~12号为只有藤络宁提取液的样品。13~17号瓶在藤络宁胶囊提取液基础上,再加入一定量的东莨菪内酯标准品。
2.2.4色谱法测定实验安捷伦1260高效液相色谱仪,FLD检测器。以ZORBAX SB-C18(4.6 mm×150 mm,5 μm)为色谱柱,流动相为甲醇-水(含0.2%磷酸)(30∶70,体积比),柱温30 ℃,流速为1.0 mL/min,检测波长为:激发波长340 nm,发射波长460 nm。
3 结果与讨论
3.1 东莨菪内酯的荧光光谱性质与影响因素
图2A为东莨菪内酯的三维荧光等高线图,可知东莨菪内酯的最大激发波长为375 nm,最大发射波长为460 nm。图2B为藤络宁胶囊的三维荧光等高线图,可知除了东莨菪内酯的光谱外,藤络宁胶囊的基体成分在此波长范围内含有与之严重重叠的高强度光谱信号,利用普通荧光法难于准确测定。故本文应用二阶校正U-PLS/RBL方法解析三维荧光光谱,以“数学分离”代替“化学分离”。
为了获得稳定和准确的分析结果,考察了溶液条件对光谱的影响。实验结果表明,东莨菪内酯在pH 7.83~11.53范围内荧光强度高且稳定,因此实验选取pH 9.0为最佳酸度条件。甲醇用量在0.1%~10%范围基本无影响,实验选择甲醇用量低于10%。实验结果表明,东莨菪内酯的质量浓度在0.014 7~0.102 9 mg/L范围内与荧光强度呈线性关系。
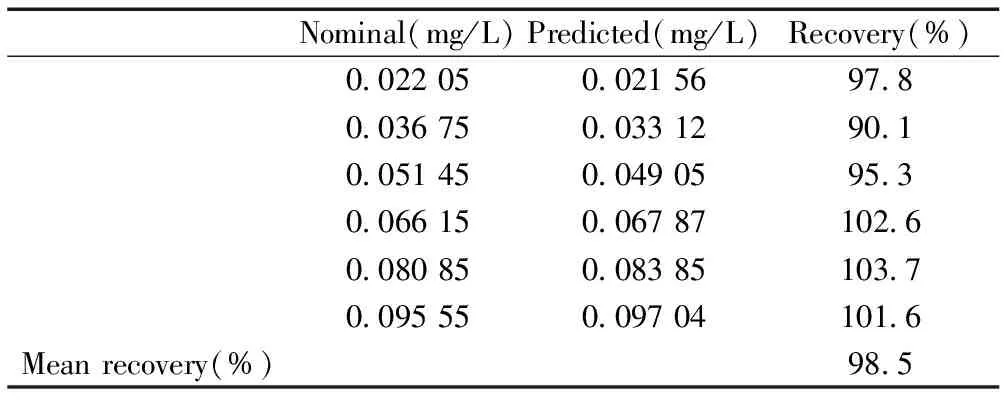
Nominal(mg/L)Predicted(mg/L)Recovery(%)0 022050 0215697 80 036750 0331290 10 051450 0490595 30 066150 06787102 60 080850 08385103 70 095550 09704101 6Meanrecovery(%)98 5
3.2 合成样的浓度解析
利用校正样进行U-PLS/RBL建模后,可对合成样进行定量分析,来验证方法的可靠性。表1为U-PLS/RBL对合成样中东莨菪内酯的定量解析结果。计算得平均回收率为98.5%,说明方法可靠。
3.3 U-PLS/RBL法解析藤络宁胶囊中东莨菪内酯含量与回收率
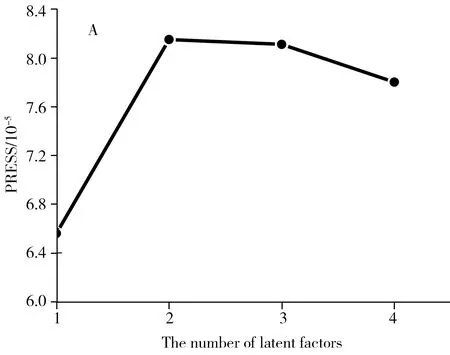
应用U-PLS/RBL首先需确定模型中的主因子数,图3为去一法得到的U-PLS/RBL的交叉验证CV图及真实浓度与预测浓度的关系曲线。从图3可知,对于东莨菪内酯的最佳因子数为1,关系曲线的相关系数为0.998,说明体系中只有1个荧光信号,这与实际情况一致。U-PLS/RBL的第二步是确定待测样中的干扰组分数,考察了U-PLS/RBL估计的光谱残差与背景组分数的变化情况,当Nunx=0时,即假定没有干扰背景,残差约为700,远大于仪器本身的噪音,说明体系中含有未知干扰组分。当背景组分≥1时,残差强度很低且接近仪器噪音,所以确定背景组分数Nunx=1。
然后利用RBL估计背景组分的激发和发射光谱,即对残差矩阵(Xu-reshape(PtRBL))进行主成分分析BRBLTRBLT后,取BRBL和TRBL矩阵的第一列。
在确定最佳因子数和背景因子数后,计算出扣除背景干扰后的藤络宁胶囊中东莨菪内酯的平均含量为0.058 9%(表2)。以色谱法测定值为标准,计算得U-PLS/RBL法解析东莨菪内酯的相对误差为3.16%,说明本方法准确。
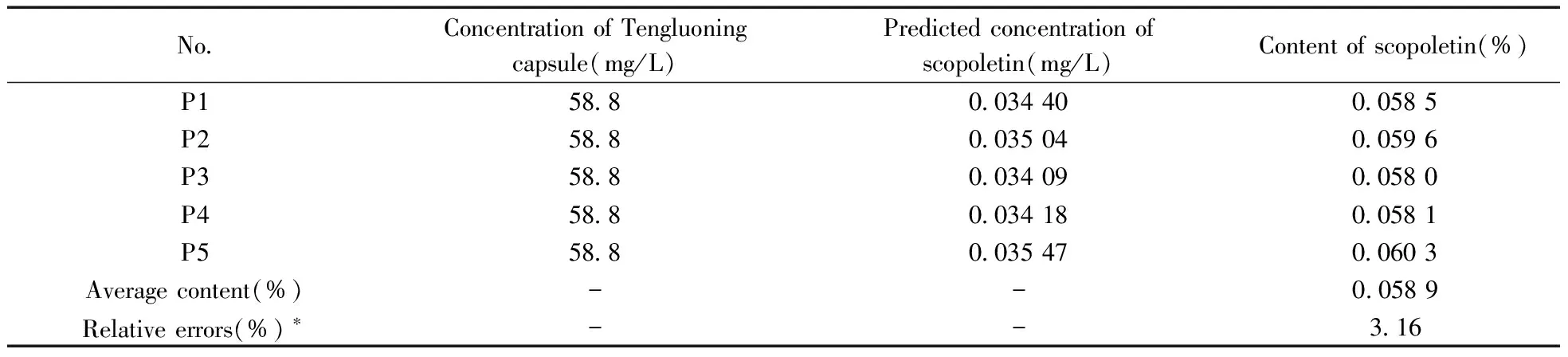
No.ConcentrationofTengluoningcapsule(mg/L)Predictedconcentrationofscopoletin(mg/L)Contentofscopoletin(%)P158 80 034400 0585P258 80 035040 0596P358 80 034090 0580P458 80 034180 0581P558 80 035470 0603Averagecontent(%)--0 0589Relativeerrors(%)∗--3 16
*the relative errors were obtained by comparing the content calculated by U-PLS/RBL with the result determined by HPLC method
用U-PLS/RBL法解析得到藤络宁胶囊中东莨菪内酯的平均回收率为100.4%,说明本方法的结果可靠。
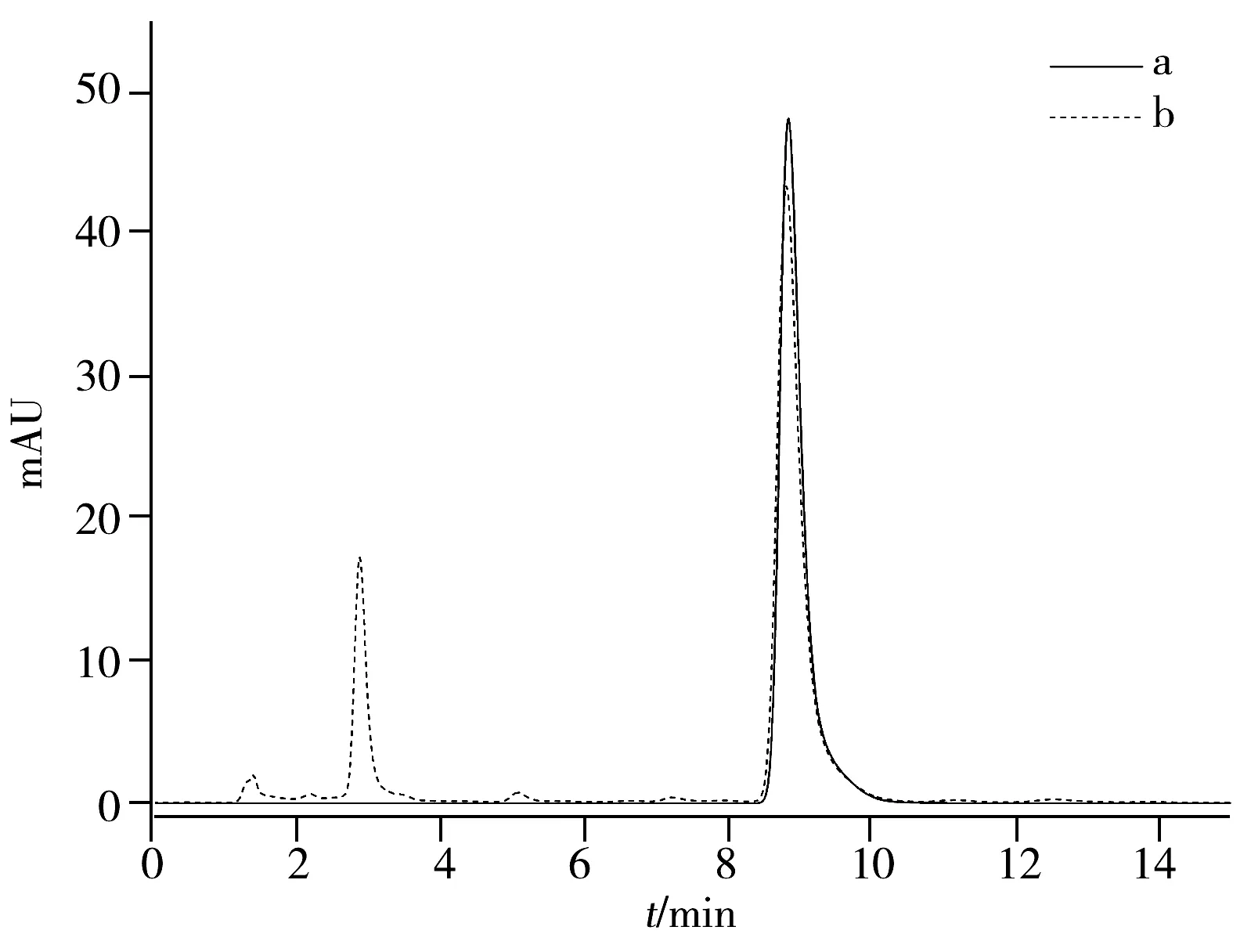
图4 东莨菪内酯(a)和藤络宁胶囊(b)的HPLC图Fig. 4 Liquid chromatograms of scopoletin(a)and Tengluoning capsule sample(b)a: 4 μL scopoletin solution(55.6 mg/L);b :5 μL Tengluoning capsule solution(19.6 g/L)
3.4 高效液相色谱法验证
在选定的最佳条件下,东莨菪内酯的保留时间为8.7 min左右(图4a)。以保留时间8.7 min的峰面积积分值(Y)为纵坐标,东莨菪内酯进样量(X,μg)为横坐标,绘制标准曲线,得回归方程:Y=102.8+1.735×104X,r=0.999 8,说明东莨菪内酯在0.029 4~0.147 μg质量范围内线性关系良好。
精密吸取对照品溶液4 μL与藤络宁胶囊样品溶液5 μL,注入高效液相色谱仪,依上述色谱条件进行分离(图4b),外标法计算含量。在东莨菪内酯的保留时间8.7 min处,根据回归方程计算得到藤络宁胶囊中东莨菪内酯的平均含量为0.057 1%(n=5,RSD=1.5%)。这与U-PLS/RBL荧光方法测得结果0.058 9%一致,证明了二阶校正方法的可靠性。
4 结 论
本文借助于荧光光谱的高灵敏性及化学计量学二阶校正方法的高选择性,成功地测定了藤络宁胶囊中东莨菪内酯的含量,与色谱法相比,本方法操作简便,绿色环保且费用较低,适合复方中药体系中有效成分含量的测定,是一种有应用潜力的方法。
参考文献:
[1] The State Drug Administration,The People′s Republic of China.Standard of Tengluoning Capsule.Standard No.WS3-642(Z-114)-2008Z(中华人民共和国国家药品监督管理局.藤络宁胶囊标准.编号:WS3-642(Z-114)-2008Z).
[2] Wu L H,Zhu E Y,Zhang Z J,Wang Z T.Chin.Tradit.HerbalDrugs(吴立宏,朱恩圆,张紫佳,王峥涛.中草药),2005,36(9):1398-1400.
[3] Song W,Jin R L,Liu J H.Chin.J.Chin.Mater.Med.(宋蔚,金蓉鸾,刘继华.中国中药杂志),1997,22(6):359-360.
[4] Chen Z,Liao L,Zhang Z,Wu L,Wang Z.J.Ethnopharmacol.,2013,150(2):501-506.
[5] Moon P D,Lee B H,Jeong H J,An H J,Park S J,Kim H R,Ko S G,Um J Y,Hong S H,Kim H M.Eur.J.Pharmacol.,2007,555:218-225.
[6] Kim H J,Jang S I,KimY J,Chung H T,Yun Y J,Kang T H,Jeong O S,Kim Y C.Fitoterapia,2004,75:261-266.
[7] Pan R,Dai Y ,Gao X,Dan L,Xia Y.Vasc.Pharmacol.,2011,54:18-28.
[8] Cao H,Qin H L,Zhong X F,Liu Y.Chin.Pharm.J.(曹红,秦红霖,钟晓峰,刘云.中国药学杂志),2001,6:411-414.
[10] Yin X L,Wu H L,Zhang X H,Gu H W,Yu R Q.AataChim.Sin.(尹小丽,吴海龙,张晓华,谷惠文,俞汝勤.化学学报),2013,71:560-566.
[11] Yang L,Liu D L,Wei Y J.J.Instrum.Anal.(杨莉,刘德龙,魏永巨.分析测试学报),2014,33(9):1038-1043.
[12] Wu H L,Li Y,Kang C,Yu R Q.Chin.J.Anal.Chem.(吴海龙,李勇,康超,俞汝勤.分析化学),2015,43(11):1038-1043.
[13] Calimag-Williams K,Knobel G,Goicoechea H C,Campiglia A D.Anal.Chim.Acta,2014,811:60-69.
[14] Bravo M A,Escandar G M,Olivieri A C,Bardin E,Aguilar L F,Quiroz W.Chemom.Intell.Lab.Syst.,2015,148:77-84.
[15] Hurtado-Sánchez M D,LozanoV A,Rodríguez-Cáceres M I,Durán-Merás I,Escandar G M.Talanta,2015,134:215-223.
[16] Alarcon F,Baez M E,Bravo M,Richter P,Escandar G M,Olivieri A C,Fuentesa E.Talanta,2013,103:361-370.
[18] Bortolato S A,Arancibia J A,Escandar G M.Environ.Sci.Technol.,2011,45:1513-1520.
[20] Haaland D M,Thomas E V.Anal.Chem.,1988,60:1193-1202.
猜你喜欢
西南大学学报(自然科学版)(2023年5期)2023-05-23 14:17:18
国学(2020年1期)2020-06-29 15:15:30
天津中医药(2020年5期)2020-06-01 12:16:14
中成药(2018年11期)2018-11-24 02:57:28
数学物理学报(2017年6期)2018-01-22 02:26:53
摄影之友(影像视觉)(2017年1期)2017-07-18 11:12:16
中成药(2017年4期)2017-05-17 06:09:27
中国民族医药杂志(2016年6期)2016-05-09 08:52:57
应用化工(2014年5期)2014-08-08 13:10:58
中成药(2014年9期)2014-02-28 22:28:56
