
17种鸡雁类IFNA的分子进化分析
2018-04-17 07:38丁恒武王青青董锦绣易双龙陈仁瑞阚显照
生物学杂志 2018年2期
丁恒武, 蒋 澜, 章 勤, 王青青, 王 莹, 董锦绣, 吴 璇, 易双龙, 陈仁瑞, 阚显照
(安徽师范大学 生命科学学院 生物信息研究所, 芜湖 241000)
干扰素(interferon,IFN)是一类在特定诱导剂(细菌、病毒等)作用下产生的一类具有抗病毒、抗肿瘤、免疫调节等生物学功能的细胞因子[1-4],由Isaacs等[5]于1957年利用鸡胚绒毛尿囊膜研究流感病毒的干扰现象时发现。根据氨基酸的序列、干扰素的功能及相应受体的结合,可将干扰素分为3种类型:Ⅰ型、Ⅱ型和Ⅲ型[6]。Ⅰ型干扰素包括IFN-α、IFN-β、IFN-ω、IFN-ε、IFN-τ、IFN-δ、IFN-κ及IFN-ξ,Ⅱ型和III型干扰素只有一种类型,分别为IFN-γ和IFN-λ[7]。其中,IFN-α(干扰素-α)由基因IFNA编码,是脊椎动物细胞受到病毒侵染时,由白细胞产生的一种调节蛋白,具有广谱的抗病毒作用[8]。各种致病性细菌或病毒对鸟类的生长繁殖影响很大,尤其是禽流感疫情带来的影响,不仅对家禽养殖业造成巨大的冲击,也对人类健康带来了巨大的隐患。已有实验表明,经过IFN-α预处理过的细胞对抵抗H7N9等禽流感病毒有重要作用[9]。
IFNA为一种重要的免疫基因,在禽类中关于其免疫功能研究已有相关报道。但关于IFNA在禽类中的分子进化研究的报道较少,因此开展禽类IFNA序列结构特征、进化选择模式和系统发育分析的研究显得非常重要。
本研究采用PCR的方法,对雁形目8种鸟类IFNA进行扩增和测序[10],并联合GenBank已释放的鸡雁小纲数据,对IFNA核苷酸序列和对应的氨基酸序列进行初步的生物信息学分析。本研究主要探讨:1)IFNA的序列结构特征;2)IFN-α的理化性质;3)IFNA编码区的进化速率;4)基于IFNA核苷酸序列的系统发育关系。
1 材料与方法
1.1 材料
本研究实验材料选取了雁形目鸭科鸟类8种,分别为赤嘴潜鸭(Nettarufina)、斑嘴鸭(Anaspoecilorhyncha)、赤麻鸭(Tadornaferruginea)、翘鼻麻鸭(Tadornatadorna)、大天鹅(Cygnuscygnus)、鸿雁(Ansercygnoides)、豆雁(Anserfabalis)、白额雁(Anseralbifrons),样本由安徽师范大学生命科学学院生物资源保护与开发省级重点实验室保存(表 1)。
本研究还选取了GenBank已释放的9种鸡雁小纲鸟类的IFNA序列作为分析材料:绿头鸭(Anasplatyrhynchos,AY879230)、灰雁(Anseranser,EU925650)、斑头雁(Anserindicus,KF731865)、疣鼻栖鸭(Cairinamoschata,JF894229)、原鸡(Gallusgallus,AM049251)、火鸡(Meleagrisgallopavo,U28140)、蓝孔雀(Pavocristatus,KJ001188)、中华鹧鸪(Francolinuspintadeanus,HM196761)和日本鹌鹑(Coturnixjaponica,AB154298)。
1.2 方法
1.2.1IFNA的序列扩增及测序
根据GenBank已释放的雁形目IFNA序列(GenBank登录号:X84764),使用在线引物设计工具Primer-BLAST(https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome)设计引物,上游引物为DNIFNA-F:5′-CCATGACCTGAAAGCGACGA-3′;下游引物为DNIFNA-R:5′-GGGCTCCGGTCAGTTCTTG-3′。
所选实验材料总DNA的提取采用酚/氯仿抽提法[11]。
以8种雁形目鸭科鸟类的总DNA为模板,进行PCR扩增。PCR扩增采用50 μL体系: ddH2O 25.94 μL,10×PCR buffer 5 μL,dNTPs(2.5 mmol/L) 6 μL,DMSO 1 μL,BSA 1 μL,Template 2 μL,DNIFNA-F(5 μmol/L) 4.4 μL,DNIFNA-R(5 μmol/L) 4.4 μL,Taqplus(5 μ/μL) 0.26 μL。PCR反应体系为:95℃预变性5 min;94℃变性30 s,55℃退火30 s,72℃延伸1 min,共36个循环;最后72℃再延伸8 min。
将PCR产物进行琼脂糖凝胶电泳检测,并将阳性扩增的产物送至生工生物工程(上海)股份有限公司进行测序。
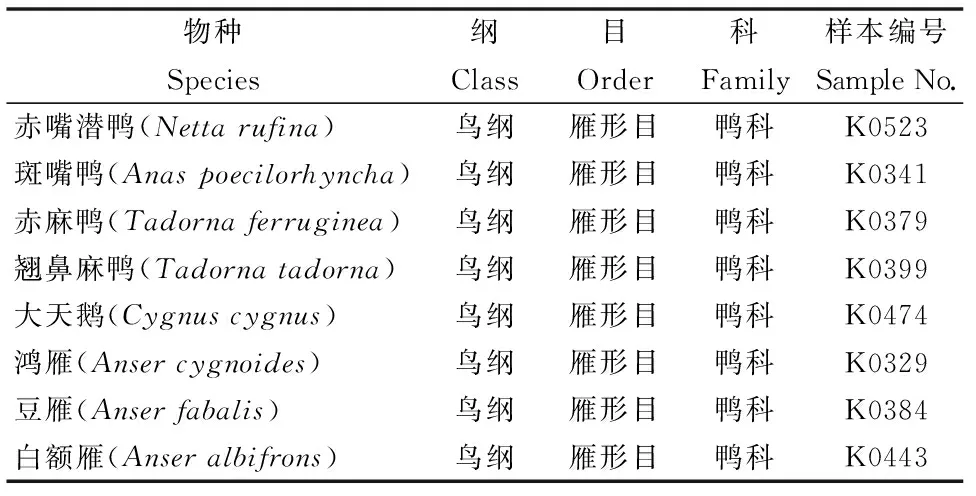
表1 研究IFNA所用到的物种来源
1.2.2IFNA的核苷酸序列分析
选取已测序列和GenBank中下载的IFNA序列,使用MEGA7.0.14[12]分析IFNA的碱基组成,并根据碱基偏选公式计算: AT Skew=(A-T)/(A+T); GC Skew=(G-C)/(G+C)。通过EMBL开发的在线工具Cpgplot分析核苷酸序列的CpG island(http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/)。
1.2.3IFNA的编码蛋白分析
IFNA编码IFN-α,蛋白质的理化性质主要利用ExPASy在线软件分析(http://web.expasy.org/protparam/);通过Signal P4.1Server软件预测IFN-α的信号肽(http://www.cbs.dtu.dk/services/SignalP/);应用TargetP 1.1工具对IFN-α的亚细胞定位进行预测(http://www.cbs.dtu.dk/services/TargetP/);通过ExPASy服务器的ProtScale程序中的K-D法,对IFN-α的疏水性/亲水性进行分析(http://web.expasy.org/protscale);通过GOR IV在线分析平台预测IFN-α的二级结构(https://npsa-prabi.ibcp.fr)。
1.2.4IFNA的进化速率分析
本研究分别选取线粒体编码氧化磷酸化复合物I、IV、V 相关基因nad2、atp6、cytb与免疫基因IFNA的进化速率分别从鸡雁小纲(17个物种)和雁形目(12个物种)进行比较。
本研究联合测序结果和GenBank中已经释放的数据(表2),采用PAML软件[13]中的位点模型对所选基因进行选择压力分析。此外本研究还采用了在线分析平台Datamonkey(http://www.datamonkey.org)中的SLAC方法对所选基因进行选择压力分析。
1.2.5系统发育树的构建
使用MAFFT version 7软件[14]进行多序列比对(http://mafft.cbrc.jp/alignment/server/),使用DAMBE version 5.3.8软件[15]对IFNA进行了饱和替代性分析,并对将多序列比对所得的矩阵用NotePad 7.3.1对序列进行拼接。
最大简约法(MP)分析,使用PAUP*4.0b10软件[16]。 采用TBR分支交换算法,自展检验1000次以确认各分支的支持率得到稳定的MP树拓扑结构。最大似然法(ML)分析,使用RaxML GUI v.1.3.1软件[17]。核苷酸替代模型采用GTRGAMMA,ML + rapid bootstrap,通过1000次自展检验评估得到稳定的ML树拓扑结构。贝叶斯法(BI)分析,使用ModelGenerator V. 0.851软件分段对核苷酸序列计算最适碱基替代模型,以随机树为起始树,4条马尔可夫链(Markov chains)运行10 000 000代(每1000代抽样1次),最后舍弃前25%的树,剩下的75%的树被用于推测支持率大于50%的一致树以及贝叶斯后验概率(Bayesian posterior probability)[18]。
本研究选择4种线粒体基因(nad2、cox1、atp6和cytb)、2种常染色体基因(IFNG和IL-2)和1种性染色体连锁基因(IFNA,位于Z染色体)来构建进化树。以企鹅目中的阿德利企鹅(Pygoscelisadeliae)作为外类群,内类群分别来自鸡雁小纲的雁形目和鸡形目[19](表2)。
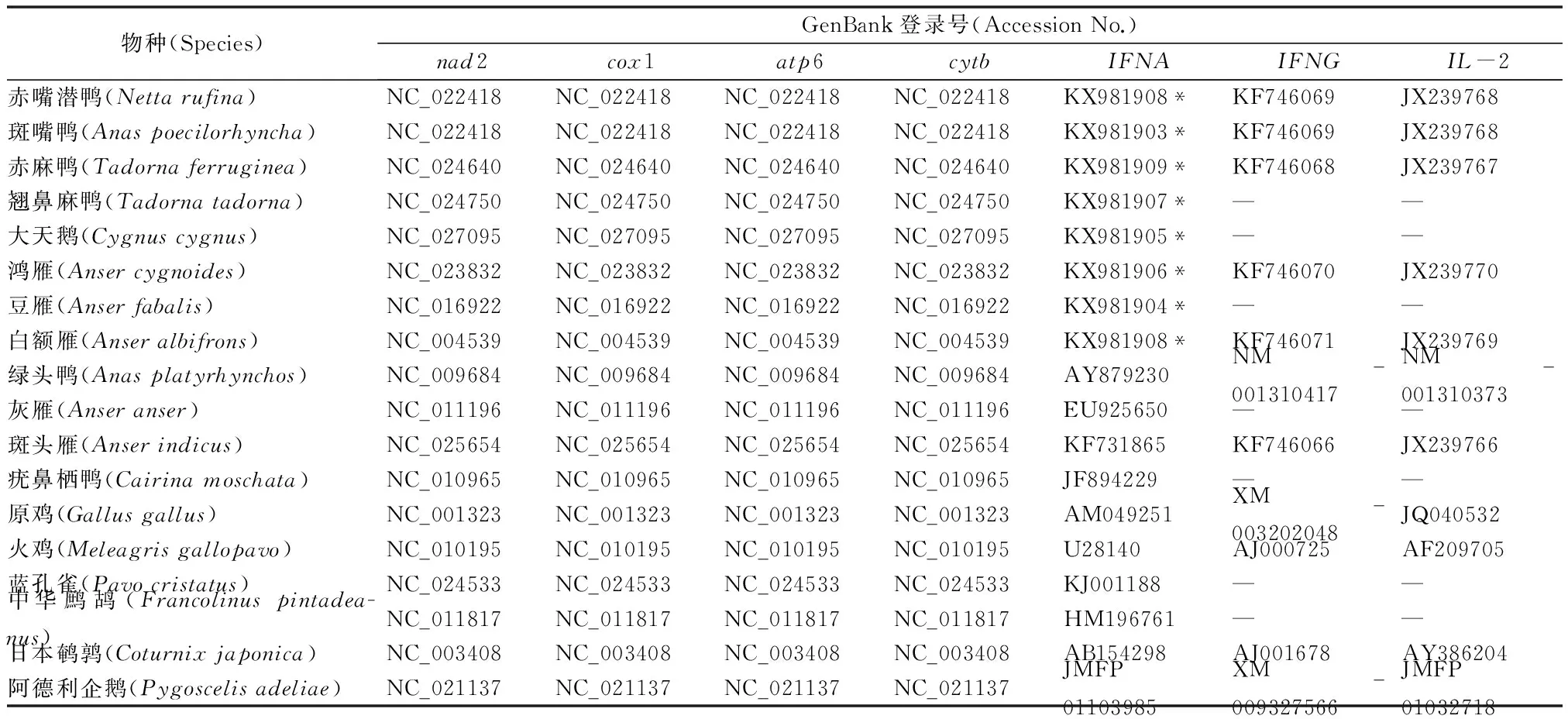
表2 本研究选择的物种及基因的GenBank登录号
* 表示该数据为本实验数据

表3 IFNA编码序列特征分析
2 结果与分析
2.1 核苷酸序列分析
本实验IFNA测序结果已递交公共基因数据库NCBI(www.ncbi.nlm.nih.gov),GenBank登录号见表2。
如表3所示,12种雁形目鸭科鸟类CDS序列长度除了赤嘴潜鸭和大天鹅为579 bp外,其他均为576 bp,GC含量在67.01%~68.92%之间,起始和终止密码子分别为ATG、TAA; 5种鸡形目雉科鸟类CDS长度在579~594 bp之间,GC含量在61.34%~63.23%之间,起始和终止密码子分别为ATG、TAG。雁形目和鸡形目鸟类均存在明显的正值AT skew与负值GC skew(图 1)。

图1 鸡雁小纲IFNA核苷酸序列的碱基偏选
上图为AT skew;下图为GC skew
所选鸡雁小纲17种鸟类中,除了蓝孔雀和中华鹧鸪IFNA核苷酸序列中不存在CpG island,其他15种鸟类均存在不同长度的CpG island,其中火鸡有2个CpG island(图2)。
2.2 IFN-α蛋白质性质分析
2.2.1IFN-α氨基酸序列比对分析
对12种雁形目鸭科鸟类和5种鸡形目雉科鸟类IFN-α氨基酸序列进行比对分析。12种雁形目鸭科鸟类IFN-α氨基酸序列长度除了赤嘴潜鸭和大天鹅为191个氨基酸外,其他均为190个氨基酸;鸡形目雉科鸟类中IFN-α氨基酸序列长度火鸡为192个氨基酸,日本鹌鹑为197个氨基酸,其他均为193个氨基酸。鸡雁小纲内IFN-α氨基酸序列矩阵为197个氨基酸残基(图3),其中66个氨基酸高度保守。
2.2.2IFN-α理化性质分析
如表4显示,在12种雁形目鸭科鸟类中,IFN-α蛋白分子质量在21.6~21.8 ku之间,等电点在7.81~9.03之间,不稳定指数在55.31~64.69之间,均为分泌通路;在5种鸡形目雉科鸟类中,IFN-α蛋白分子质量在21.9~22.3 ku之间,等电点在8.28~9.08之间,不稳定指数在65.21~80.44,均为分泌通路;鸡雁小纲17个物种的亲水性平均系数均为负值,说明IFN-α蛋白是可溶性蛋白。
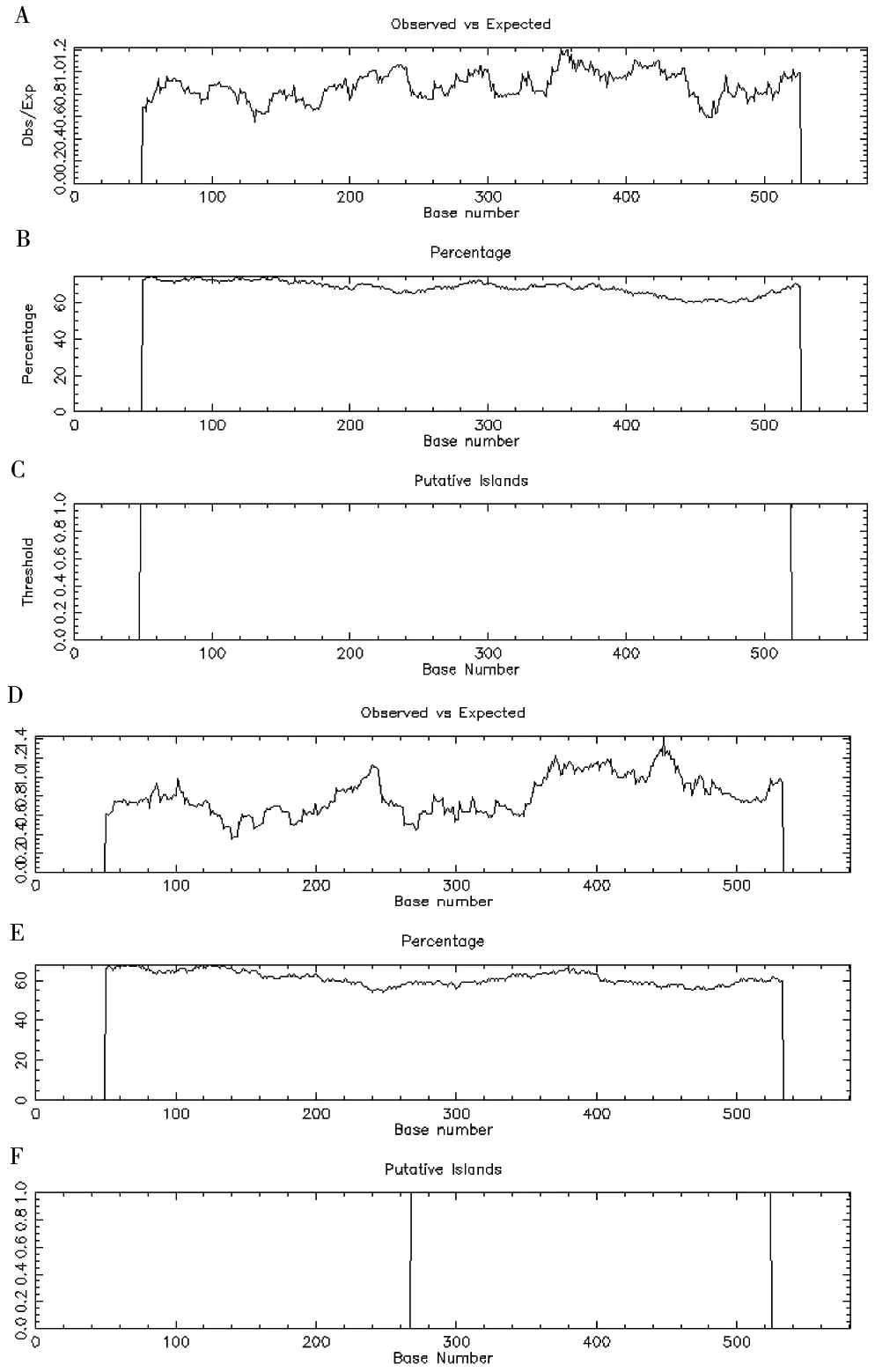
图2 IFNA CpG island预测
A~C为鸿雁;D~F为原鸡的IFNACpG island。图中Percentage表示C%+G%,Obs/Exp表示观测到的CG与CG%的比值,Putative Islands表示综合预测的CpG island的位置
将所选IFN-α氨基酸序列用TargetP工具分析显示,在12种雁形目鸭科鸟类中IFN-α除大天鹅、赤嘴潜鸭编码的IFN-α信号肽剪切位点位于氨基酸序列29位与30位之间,其他物种蛋白信号肽剪切位点位于氨基酸序列28位与29位之间;在5种鸡形目雉科鸟类中,原鸡、蓝孔雀、中华鹧鸪信号肽剪切位点位于氨基酸序列31位与32位之间,火鸡信号肽剪切位点位于氨基酸序列30位与31位之间,日本鹌鹑信号肽剪切位点位于氨基酸序列25位与26位之间。图4显示的是鸿雁和原鸡的信号肽预测。

图3 干扰素-α氨基酸序列比对Fig 3 Amino acid sequence alignment of IFN-α
2.2.3IFN-α的二级结构预测
IFN-α的二级结构包括无规则卷曲、5个α-螺旋、伸展链。12种雁形目鸟类IFN-α无规则卷曲占52.15%~61.96%,α-螺旋占28.22%~38.04 %,伸展链占9.82%~12.27%;5种鸡形目鸟类IFN-α无规则卷曲占53.89%~58.64%,α-螺旋占26.54%~37.65%,伸展链占6.17%~14.81%(表5)。

图4 IFNA信号肽预测
上图为鸿雁;下图为原鸡的信号肽预测。C值表示剪切位点分值,S值表示信号肽分值,Y值表示综合剪切点分值
2.3 IFNA进化速率分析
现在常用dN/dS描述基因的进化速率,如表6所示。在鸡雁小纲内,17种鸟类基因atp6、cytb、nad2的dN/dS值均小于IFNA的dN/dS值,说明所选线粒体基因进化速率小于免疫基因IFNA。在雁形目内,12种鸟类所选的基因也出现类似的情况。使用PAML位点特异性模型M0(单一速率)和M3(离散的速率)的比较来评判位点间选择压力未检测到正选择位点;使用Datamonkey的SLAC算法,在鸡雁小纲水平,免疫基因IFNA找到1个正选择位点:Codon 16(P=0.0115)。
2.4 系统发育分析
经过多序列比对,我们得到了以下序列矩阵:1)nad2编码区长度为1041 bp;2)cytb编码区长度为1143 bp;3)cox1编码区长度为1551 bp;4)atp6编码区长度为684 bp;5)IFNA编码区长度为558 bp;6)IFNG编码区长度为498 bp;7)IL-2编码区长度为429 bp。

表6 基因进化速率分析
上述7个序列矩阵的最适碱基替代模型:1)nad2最适模型为TrN+G;2)cytb最适模型为K81uf+G;3)cox1最适模型为GTR+I;4)atp6最适模型为HKY+G;5)IFNA最适模型为TrN;6)IFNG最适模型为HKY+I;7)IL-2最适模型为HKY+G。
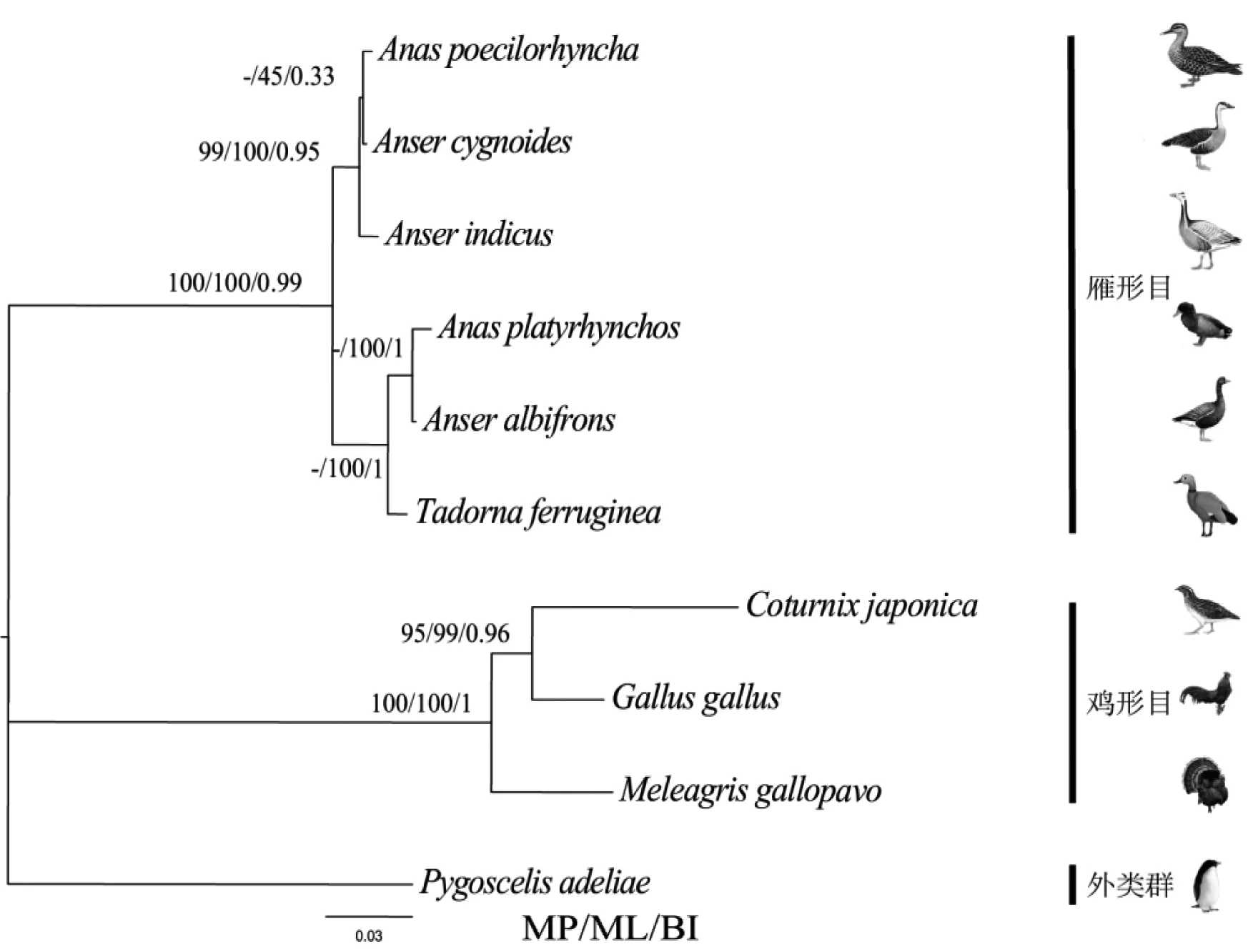
本研究中系统发育树图的枝长和拓扑结构源于贝叶斯推断法树图,并分别用MP、ML法数据标注支持率,BI法数据标注后验概率。本研究先后构建基于IFNA单基因的系统发育树和基于上述7种基因联合构建的系统发育树。
饱和替代性分析结果是Iss=6608 表4 干扰素-α的氨基酸理化性质的比较 表5 IFN-α的二级结构预测 图5基于IFNA序列构建的系统发育树 Fig 5 Phylogenetic trees based on sequences ofIFNA 通过nad2、cytb、cox1、atp6、IFNA、IFNG和IL-2 7种基因联合构建的系统发育树显示(图6),鸡形目3种鸟类聚成一大枝,ML和MP的自展检验值均为100,BI后验概率为1,鸡形目3种鸟构成很强的单系;雁形目6种鸟类聚成一大枝,ML和MP的自展检验值均为100,BI后验概率为0.99。雁形目鸭科不同属的物种的系统发育关系较为稳定,有着很高的支持率和后验概率:Tadorna属与Anas属具为一支(ML和MP的自展检验值均为100,BI后验概率为0.99)与Anser属互为姐妹群(ML和MP的自展检验值均为100,BI后验概率为0.99),多基因联合构建的系统发育树可以较好地反映所选物种进化的历史。 鸡雁小纲IFNA核苷酸序列较高的负值GC skew。其原因是IFNA编码序列中高比例的C碱基,其中雁形目鸭科鸟类C>49.0%,鸡形目雉科鸟类C>41.8%。通过在线工具Cpgplot证实了我们的发现。CpG island说明这段序列是含有丰富的5-甲基胞嘧啶,即可能存在DNA甲基化。 图 6 基于多基因序列构建的系统发育树 干扰素-α蛋白分子质量在21.6~22.3 ku之间,等电点在7.81~9.08之间,不稳定指数在55.31~80.44之间,均为亲水性分泌蛋白,存在信号肽及其切割位点,根据Guruprasad方法[20]表明当不稳定系数分值大于40时,预测蛋白质不稳定,故干扰素-α蛋白为不稳定蛋白。本研究发现,所选物种中雁形目鸭科鸟类干扰素-α蛋白的分子质量和不稳定系数普遍小于鸡形目稚科干扰素-α蛋白的分子质量和不稳定系数。 非同义替换(dN)指基因编码的氨基酸从一种替换成另一种的现象;同义替换(dS)指基因编码的氨基酸不改变的替换。如果dN/dS=1,则基因受中性进化,dN/dS<1时,则基因受纯化选择,dN/dS>1,则认为基因受正选择,本研究中所选的基因均为dN/dS<1,故它们都受纯化选择作用。通常情况下,线粒体基因进化速率要快于核基因[21]。在本研究中分析dN/dS时,发现所选物种无论是在鸡雁小纲内还是在雁形目内,它们细胞核上免疫相关基因IFNA的进化速率都快于线粒体基因nad2、cytb及atp6。并且在鸡雁小纲中IFNA发现了1个正选择位点,说明在雁形目和鸡形目两大类群的分歧过程中,IFNA可能经历了功能适应性进化。 由于线粒体基因严格的母系遗传、进化速率稳定等优点[21-22],使得线粒体基因成为研究进化的好材料,将线粒体基因加入到Z染色体连锁基因或常染色体基因构建联合系统发育树,结果将会更加可靠[19]。通过构建系统发育树,本研究发现所选物种中,IFNA单基因不能构建稳定的系统发育树,当Z染色体连锁基因IFNA与常染色体基因IFNG、IL-2以及线粒体基因nad2、cox1、atp6和cytb联合构建系统发育树时可以得到稳定的、支持率高和后验概率高的系统发育树,这说明IFNA基因携带系统发育信息。 本次研究通过PCR测序并联合已释放的数据,通过生物信息学分析,探讨了鸡雁小纲IFNA的序列特征、进化速率、系统发育意义以及其编码蛋白IFN-α的性质。为进一步探讨鸟类IFNA的分子进化机制和生物学功能奠定了基础。 [1]LITVIN O, SCHWARTZ S, WAN Z, et al. Interferon α/β enhances the cytotoxic response of MEK inhibition in melanoma[J]. Molecular Cell, 2015, 57(5): 784-796. [2]MORO K, KABATA H, TANABE M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses[J]. Nature Immunology, 2015, 17(1): 76-86. [3]NICE T J, BALDRIDGE M T, MCCUNE B T, et al. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity[J]. Science, 2015, 347(6219): 269-273. [4]YAMADA T, HORIMOTO H, KAMEYAMA T, et al. Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense[J]. Nature Immunology, 2016, 17(6): 687-694. [5]ISAACS A, LINDENMANN J. Virus interference. I. The interferon[J]. Proceedings of the Royal Society of London, 1957, 147(927): 258-267. [6]SHEPPARD P, KINDSVOGEL W, XU W, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R[J]. Nature Immunology, 2003, 4(1): 63-68. [7]XU L, YANG L, LIU W. Distinct evolution process among type I interferon in mammals[J]. Protein Cell, 2013, 4(5): 383-392. [8]HOLKO M, WILLIAMS B R. Functional annotation of IFN-alpha-stimulated gene expression profiles from sensitive and resistant renal cell carcinoma cell lines[J]. Journal of Interferon & Cytokine Research, 2006, 26(8): 534-547. [9]ARILAHTI V, MKELS M, TYNELL J, et al. Novel avian influenza A(H7N9) virus induces impaired interferon responses in human dendritic cells[J]. PLos One, 2014, 9(5): e96350. [10]LOWENTHAL J W, STAEHELI P, SCHULTZ U, et al. Nomenclature of avian interferon proteins[J]. Journal of Interferon & Cytokine Research the Official Journal of the International Society for Interferon & Cytokine Research, 2001, 21(7): 547-549. [11]SAMBROOK J, RUSSELL D W. Molecular cloning: a laboratory manual. 3rd edn[M]. New York:Cold Spring Harbor Laboratory Press, 2001. [12]KUMAR S, STECHER G, TAMURA K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets[J]. Mol Biol Evol, 2016, 33(7): 1870-1874. [13]YANG Z. PAML: a program package for phylogenetic analysis by maximum likelihood[J]. Bioinformatics, 1997, 13(5): 555-556. [14]KATOH K, STANDLEY D M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability[J]. Molecular Biology & Evolution, 2013, 30(4): 772-780. [15]XIA X. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution[J]. Molecular Biology & Evolution, 2013, 30(7): 1720-1728. [16]SWOFFORD D L. Phylogenetic analysis using parsimony[J]. Sunderland, MA: Sinauer Associates Inc, software, 2002. [17]SILVESTRO D, MICHALAK I. raxmlGUI: a graphical front-end for RAxML[J]. Organisms Diversity & Evolution, 2012, 12(4): 335-337. [18]JIANG L, CHEN J, WANG P, et al. Correction: the mitochondrial genomes ofAquilafasciataandButeolagopus(Aves, Accipitriformes): sequence, structure and phylogenetic analyses[J]. Plos One, 2015, 10(10): e0141037. [19]CORL A, ELLEGREN H. Sampling strategies for species trees: the effects on phylogenetic inference of the number of genes, number of individuals, and whether loci are mitochondrial, sex-linked, or autosomal[J]. Molecular Phylogenetics & Evolution, 2013, 67(2): 358-366. [20]GURUPRASAD K, REDDY B V, PANDIT M W. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence[J]. Protein Engineering, 1990, 4(2): 155-161. [21]BOORE J L. Animal mitochondrial genomes[J]. Nucleic Acids Research, 1999, 27(8): 1767-1780. [22]SOLIGNAC M, MONNEROT M, MOUNOLOU J C. Mitochondrial DNA heteroplasmy inDrosophilamauritiana[J]. Proceedings of the National Academy of Sciences of the United States of America, 1983, 80(22): 6942-6946.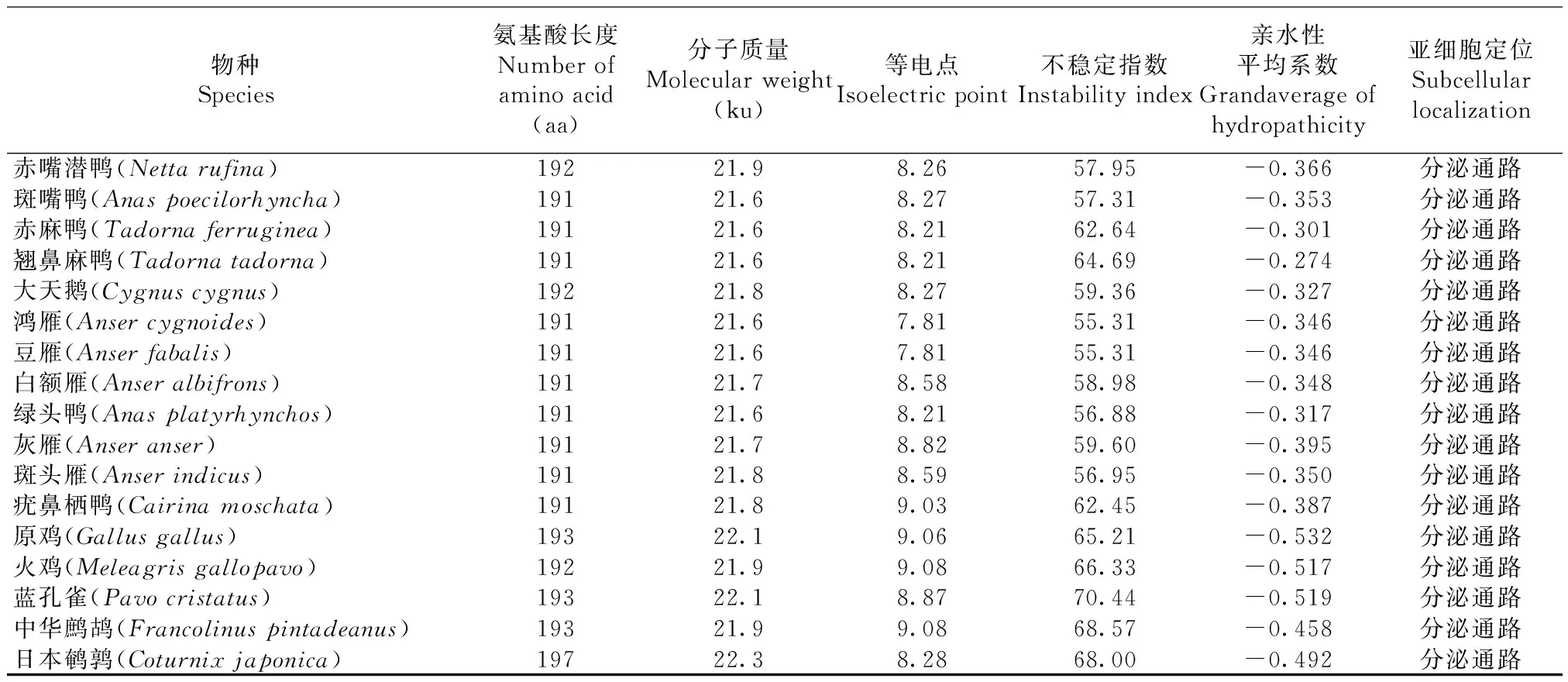

3 讨论

猜你喜欢
学与玩(2022年9期)2022-10-31
生物技术通报(2021年6期)2021-08-11
文苑(2020年12期)2020-04-13
小太阳画报(2019年1期)2019-06-11
中国洗涤用品工业(2019年4期)2019-05-11
中成药(2018年1期)2018-02-02
小学生必读(低年级版)(2017年5期)2017-08-12
中成药(2017年3期)2017-05-17
浙江农业学报(2017年3期)2017-04-08
中国烟草学报(2016年1期)2016-11-16
