
二氢生物喋呤还原酶减轻脂肪酸诱导的细胞凋亡的机制研究
2018-03-30 03:03古艳婷孙斯凡
安徽医科大学学报 2018年2期
古艳婷,冯 茹,孙斯凡
糖尿病肾病是由很多因素导致的疾病,患者体内存在多种通路的失调,至今仍未完全阐明。研究显示糖尿病肾病存在自噬现象的下调,其中肾脏中的脂代谢紊乱会促进肾小球硬化和肾小管间质纤维化[1-2],且会导致肾小管细胞凋亡[3]。研究者在前期实验中发现,二氢生物喋呤还原酶(dihydropteridine reductase,QDPR)在体外高表达可抑制细胞的氧化应激,但其对活性氧簇(reactive oxygen species,ROS)的抑制是否参与糖尿病肾病的发生发展还不确定[4]。本研究构建了野生型QDPR重组质粒并转染至HEK293T细胞,使用脂肪酸(plamitic acid, PA)刺激细胞,观察QDPR过表达对PA诱导的细胞凋亡作用的影响,探讨QDPR在糖尿病肾病发病机制中的作用。
1 材料与方法
1.1主要试剂及细胞鼠单抗V5抗体(美国Invitrogen公司);兔多抗LC3(美国Sigma公司),兔多抗Beclin 1抗体(美国Sigma-Aldrich公司),二抗(美国Abcam公司);凝胶回收纯化试剂盒、质粒中提试剂盒(德国QIAGEN公司);限制性内切酶EcoR V、Pst I、Xba I(美国NewEngland Biolabs公司);四氢生物喋呤(tetrahydrobiopterin,BH4)检测试剂盒(北京antibody-online公司),ROS检测试剂盒(北京Applygen有限公司)。HEK293T细胞为航空总医院中心实验室保存。
1.2QDPR重组质粒的构建在大鼠QDPR引物上游和下游分别引入EcoR V酶切位点和 Xba I 酶切位点。引物序列如下:上游5′-AGATATCTGGCGGCTTCGGGCGAGGC-3′;下游5′-ATCTAGAGAAATAGGCTGGAGTAAGCT-3′,PCR反应条件为:94 ℃预变性2 min;94 ℃变性30 s;54 ℃退火30 s;72 ℃延伸1 min。经双酶切后再使用T4 DNA连接酶连接目的片段与载体,构建rQDPR/pcDNA3.1/V5-His重组质粒。
1.3HEK293T细胞转染及分组实验分为A、B、C 3组,A组为空载体组, B组为经过PA刺激的空载体转染组,C组为经过PA刺激的QDPR重组质粒转染组。HEK293T细胞常规培养于含10%胎牛血清的DMEM培养基中,转染前24 h传代,以2×106/孔的细胞密度接种于6孔板中,在37 ℃、5% CO2条件下培养24 h,细胞贴壁后利用磷酸钙共沉淀法转染。转染24 h后,空载体组换成正常培养基,B组和C组加入含浓度为0.5 mmol/L PA的培养基。培养24 h后收集细胞液和细胞进行检测。
1.4BH4和ROS生成量检测使用BH4试剂盒检测细胞和细胞液中BH4的含量。在细胞中加入含有1 mmol/L DCFH-DA的培养基,然后在37 ℃条件下孵育30 min,随后用PBS清洗2次,使用荧光显微镜观察并计数。
1.5Westernblot检测各组取60 μg蛋白样品进行SDS-PAGE电泳并电转移至PVDF膜,5 g/L脱脂奶粉封闭2 h,TBST洗膜,分别用鼠单抗V5抗体 (1 ∶5 000)、Caspase 3抗体(1 ∶2000)、 Beclin1抗体(1 ∶2 000)、Beclin2抗体(1 ∶2 000)及β-actin 抗体(1 ∶2 000) 4 ℃孵育过夜;使用浓度为1 ∶4 000的二抗孵育1 h,TBST洗膜3次后ECL显影。采用Image J软件分析条带光密度值, 并计算各指标与β-actin光密度值的比值。
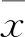
2 结果
2.1QDPR融合蛋白成功表达用鼠单抗V5抗体检测到C组QDPR融合蛋白的表达,A组以及B组未见目的蛋白表达,见图1。

图1 Western blot检测转染后QDPR融合蛋白表达
2.2QDPR过表达对BH4含量的影响与B组相比,C组细胞和细胞液中BH4的含量明显增多(P<0.05),A组以及B组之间BH4的生成量差异无统计学意义。见图2。
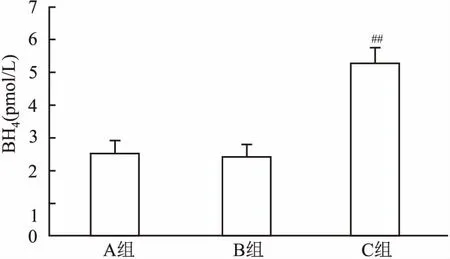
图2 QDPR过表达对BH4含量的影响与B组比较:##P<0.01
2.3QDPR过表达对PA诱导的ROS生成的影响PA刺激HEK293T细胞后,B组内ROS生成增多,QDPR过表达(C组)显著减少PA诱导的ROS生成,三组间F=263.95,可见QDPR显著降低ROS的生成。见图3。

图3 QDPR过表达对PA诱导的ROS生成的影响与A组比较:**P<0.01;与B组比较:##P<0.01
2.4QDPR过表达对PA诱导的细胞凋亡通路分子改变的作用经过0.5 mmol/L PA刺激细胞24 h后,Western blot法检测结果显示:与A组相比,B组经过PA刺激后细胞中Beclin 1和Caspase 3表达水平显著上调(P<0.05),Beclin 2表达水平下降(P<0.05);QDPR过表达后,与B组比较,C组Beclin 1和Caspase 3表达水平显著下调(P<0.05),Beclin 2表达水平升高 (P<0.05)。见图4。

图4 QDPR过表达对细胞凋亡通路分子蛋白表达的影响±s)
3 讨论
QDPR在BH4再生过程中起到重要作用,并对维持体内BH4含量稳定有很重要意义[5]。而BH4是三种一氧化氮合酶(nitricoxide synthase,NOS)激活所必需的辅助因子,当BH4在体内的含量不足时,NOS会产生脱偶联导致ROS生成增多[6]。ROS又与糖尿病肾病的发生关系密切[7]。所以笔者提出假设QDPR可能通过BH4来调节ROS的含量,继而影响细胞的自噬功能。课题组在前期差异蛋白质组学研究[8]中发现,在糖尿病肾病发展过程中,QDPR发生了突变,并且可以调节氧化应激过程与肾脏纤维化过程。但QDPR是否参与细胞凋亡的发生发展有待进一步研究。
糖尿病肾病患者的体内存在明显的PA紊乱,糖尿病肾病患者体内PA增高会导致氧化应激、炎症等通路的激活,这些通路的激活都参与了糖尿病肾病的发生发展[9],高浓度的PA在肾脏中聚集还能诱导肾小管上皮细胞产生自噬并发生凋亡[10-11]。凋亡是由十分复杂的信号转导通路调控的,细胞凋亡的分子机制中重要的效应因子是Caspase家族。Caspase 3是评价哺乳动物凋亡的关键蛋白酶,它降解凋亡过程中标志性底物的活性最强,同时,也可以通过调节下游分子的表达来介导细胞的凋亡[12]。此外,抑制凋亡的家族成员(Bcl-2)是主要抗凋亡因子[13-14]。近年来有研究[15]显示在链脲佐菌素诱导的1型糖尿病肾病大鼠肾组织中存在着细胞凋亡活性的下调,包括促细胞凋亡基因表达较少以及抑制细胞凋亡基因表达增加。Beclin家族中的Beclin-2可以防止凋亡产生,是体内调控细胞凋亡的基因之一,它通过调节凋亡相关蛋白来达到抑制凋亡的目的。而Beclin 1是作为Beclin 2的交互蛋白被分离出来的,Beclin 1拥有与Beclin 2相似的结构域,之后发现Beclin 1有促凋亡的作用,并且随着细胞内ROS的生成,和Vps-Beclin 1复合体有关的自噬也被启动,从而引起凋亡[16]。本研究结果显示转染野生型QDPR重组质粒后细胞中Beclin 2蛋白水平显著增高,Beclin 1和Caspase 3的表达明显降低,提示QDPR可能有诱导细胞凋亡的作用,由此推测QDPR可能和糖尿病肾病的发生发展有关,并且课题组发现的QDPR可能是通过氧化应激通路来调节细胞凋亡,但是它的具体分子机制还不清楚,有待进一步研究。
[1] Chen H C,Guh J Y,Chang J M,et al.Role of lipid control in diabetic nephropathy[J].Kidney Int Suppl, 2005, 67(94):S60-2.
[2] Tolonen,N,Forsblom C,Thorn L,et al.Lipid abnormalities predict progression of renal disease in patients with type 1 diabetes[J].Diabetologia, 2009, 52(12):2522-30.
[3] Wlodkowic D,Telford W,Skommer J,et al.Apoptosis and beyond:cytometry in studies of programmed cell death[J].Methods Cell Biol, 2011, 103(6):55-98.
[4] Gu Y, Gong Y, Zhang H, et al. Regulation of transforming growth factor beta1 gene expression by dihydropteridine reductase in kidney 293T cells[J]. Biochem Cell Biol, 2013, 91(1): 187-93.
[5] Wang C C,Liu T Y,Cheng C H,et al.Involvement of the mitochondnon-dependent pathway and oxidative stress in the apoptosis of murine splenocytes induced by areca nut extract[J].ToxicolInVitro, 2009, 23(5):840-7.
[6] Sarkar S, Korolchuk V, Renna M, et al. Complex inhibitory effects of nitric oxide on autophagy[J]. Mol Cell, 2011, 43(1): 19-32.
[7] Song C, Mitter S K, Qi X, et al. Oxidative stress-mediated NFκB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy[J]. PLoS One, 2017, 21(2): 39527-33.
[8] 古艳婷, 赵婷婷, 李 平, 等. 二氢生物喋呤还原酶参与调控HEK293T细胞自噬作用的初步研究[J]. 中国比较医学杂志, 2012, 22(11):1-5.
[9] 成 芳,涂珍珍,周利利,等. 雷帕霉素对肾小管上皮细胞生物学行为的影响及其治疗糖尿病肾病分子机制的初步研究[J]. 安徽医科大学学报, 2017,52(1):1-6.
[10] Rosca M G,Vazquez E J,Chen Q,et al.Oxidation offatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes[J].Diabetes, 2012, 61(8):2074-83.
[11] Wlodkowic D,Telford W,Skommer J,et al.Apoptosis and beyond:cytometry in studies ofprogrammed cell death[J].Methods Cell Biol, 2011, 103(4):55-98.
[12] Wu W H,Zhang, M P,Zhang F,et al. The role of programmed cell death in streptozotocin-induced early diabetic nephropathy[J]. J Endocrinol Invest, 2011, 34(9):e296-301.
[13] Chew E Y, Ambrosius W T, Davis M D, et al.Effects of medical therapies on retinopathy progression in type 2 diabetes[J].N Engl J Med, 2010, 363:233-44.
[14] 罗红波,杨金升,石向群,等. Aβ诱导内质网应激性凋亡通路的启动及二苯乙烯苷的影响[J]. 东南大学学报:医学版,2011,30( 6): 855-60.
[15] Hewitt G, Korolchuk V I. Repair, Reuse, Recycle: the expanding role of autophagy in genome maintenance[J]. Trends Cell Biol, 2017, 27(5):340-51.
[16] Borutaite V,Jekabsone A,Morkuniene R,et al.Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction,cytochrome c telease and apoptosis induced by heart ischemia[J].J Mol Cell Cardiol, 2003, 35(4):357-66.
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
陕西中医药大学学报(2022年4期)2022-08-01
湖南畜牧兽医(2021年6期)2022-01-24
食品安全导刊(2021年21期)2021-08-30
中老年保健(2021年4期)2021-08-22
中老年保健(2021年4期)2021-08-22
猪业科学(2021年5期)2021-06-02
中国畜禽种业(2021年4期)2021-05-21
江西农业学报(2021年4期)2021-04-20
