
五加皮酒质量标准的提高
2018-02-28 11:12吴晓龙
中医研究 2018年2期
刘 杰,吴晓龙
(1.河南省中医药研究院,河南 郑州 450004; 2.河南省汉帝药业有限公司,河南 平顶山 467000)
五加皮酒由五加皮、陈皮、当归、川芎等26味中药组成,具有舒筋活血、除湿祛风功能,用于风湿痹痛,手足痉挛,四肢麻木,腰膝酸痛[1]。原质量标准仅有性状和常规检查项目,不能满足质量控制的要求。本研究增加了五加皮酒中当归、川芎和独活的薄层色谱鉴别法和主要成分橙皮苷的高效液相色谱含量测定法,以提高其质量标准,更好地控制本品质量。
1 试药与仪器
五加皮酒,由河南省汉帝药业有限公司提供,批号20170101,20170102,20170103。橙皮苷,含量测定用,由中国药品生物制品鉴定所提供,批号0721-200010。色谱纯级甲醇;硅胶G,青岛海洋化工厂产品。所用其他试剂均为分析纯。Waters 2695高效液相色谱仪,美国Waters公司产品,配备Waters 2696二极管阵列检测器,Empower色谱工作站;C-18柱(250 mm id×4.6mm,5μm,美国热电公司产品)。
2 方法与结果
2.1 定性鉴别
2.1.1 当归和川芎的薄层色谱鉴别
按照《中国药典》2015版通则0502薄层色谱法试验。取本品30 mL,用乙醚振摇提取2次(25,20 mL),合并乙醚液,挥干,残渣加甲醇2 mL使溶解,转入小容量瓶,得供试品溶液。取当归、川芎对照药材各0.5 g,分别加乙醚10 mL,超声处理5 min,滤过,挥干,残渣加甲醇2 mL使溶解,转入小容量瓶,得对照药材溶液。在同一用5 g/L羧甲基纤维素钠水溶液制备的硅胶G薄层板上,分别点样供试品溶液10 μL、当归和川芎对照药材溶液各5 μL。展开剂为乙酸乙酯-正己烷(1∶9),展开,晾干,于紫外光灯(365 nm)下检视。结果:供试品色谱中,在与2种对照药材色谱相应的位置上显相同颜色的荧光斑点。另取当归和川芎阴性对照样品,同法进行试验。结果:当归和川芎阴性对照样品色谱中,在与两种对照药材色谱相应的位置上没有相应的斑点。结果表明:本法专属性高。见图1。

1~3.3批样品;4.当归对照药材;5.川芎对照药材;6.当归和川芎阴性对照品图1 当归和川芎的薄层色谱鉴别
2.1.2 独活的鉴别
按《中国药典》2015版通则0502薄层色谱法试验。供试品溶液采用2.1.1项下的同一种溶液。取蛇床子素对照品置小量瓶中,加甲醇使溶解,制成浓度为0.2 g/L的溶液作为对照品溶液。另取独活对照药材0.5 g,加甲醇10 mL,超声提取10 min,滤过,滤液作为对照药材溶液。在同一用5 g/L羧甲基纤维素钠水溶液制备的硅胶G薄层板上,分别点样供试品溶液10 μL、对照品溶液和对照药材溶液各5 μL,展开剂为乙酸乙酯-石油醚(60~90 ℃) (3∶7),展开后晾干,于紫外光灯(365 nm)下检视。结果:供试品色谱中,在与对照品色谱和对照药材色谱相应的位置上,分别显相同颜色的荧光斑点。另取独活阴性对照品同板进行试验,结果:阴性对照品的薄层色谱中,在与蛇床子素对照品和独活对照药材薄层色谱相应的位置上,没有相同颜色的荧光斑点。结果表明:该试验方法专属性强,见图2。

1~3.3批样品;4.独活对照药材; 5.蛇床子素对照品;6.独活阴性对照品图2 独活的薄层色谱鉴别
2.2 含量测定
2.2.1 制备对照品溶液
取橙皮苷对照品适量,置25 mL容量瓶中,精密称定为7.30 mg,加甲醇适量,超声处理5 min使溶解,加甲醇稀释至刻度,摇匀。精密量取2 mL,转入10 mL量瓶中,加甲醇2 mL稀释,再加水至刻度,摇匀,即得浓度为58.40 μg/L的橙皮苷对照品溶液。
2.2.2 制备供试品溶液
取本品滤过,弃去初滤液,取续滤液,即得。
2.2.3 测定波长的选择
对橙皮苷对照品色谱峰和样品色谱中相应的色谱峰提取二极管阵列检测器光谱,结果:二者的紫外光谱吸收基本一致,均在285 nm处有强吸收,与《中国药典》2015版陈皮项下规定的波长基本一致。故采用测定波长为283 nm。
2.2.4 色谱条件
以十八烷基硅烷键合硅胶为色谱柱填充剂;检测波长283 nm;流动相为甲醇-20 g/L乙酸水溶液(40∶60)。按《中国药典》规定,理论板数按橙皮苷峰计算应不低于2 000。
曾对比甲醇-20 g/L乙酸水溶液(37∶63)、甲醇-20 g/L 乙酸水溶液(42∶58)和甲醇-20 g/L乙酸水溶液(40∶60)3种流动相。结果:以甲醇-20 g/L乙酸水溶液(40∶60)色谱的保留时间和分离效果更满意,故采用该流动相。
在此色谱条件下,分别精密吸取对照品溶液和供试品溶液各10 μL,注入液相色谱仪,测定,计算。
2.2.5 标准曲线的绘制
取对照品溶液,依次精密进样2,5,10,15,20,25 μL,测定橙皮苷成分的峰面积。以进样量为横坐标x,以峰面积为纵坐标y,绘制标准曲线。测得橙皮苷的标准曲线为:Y=1.314×103X-3.387×104,r=0.999 9。结果表明:橙皮苷进样量在 0.116 8~1.460 0 μg之间有良好的线性关系,直线近乎过原点。因此,可以采用外标一点法进行含量测定。
2.2.6 阴性对照试验
按制备工艺和处方比例,称取除陈皮以外的各味药材,制得陈皮阴性对照样品。按供试品溶液制备方法,制得陈皮阴性对照溶液。分别吸取供试品溶液、橙皮苷对照品溶液和阴性对照溶液各10 μL,进样测定。结果:供试品色谱中在与对照品色谱相对应的保留时间有色谱峰出现,而阴性对照溶液在相对应位置无相应的色谱峰。结果表明:本法专属性强。见图3。
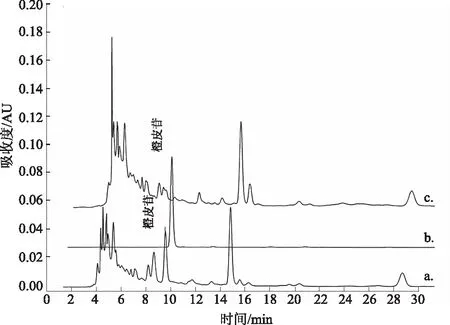
a.五加皮酒;b.橙皮苷对照品;c.陈皮阴性对照品图3 橙皮苷测定的HPLC色谱图
2.2.7 进样精密度试验
取同一供试品溶液,按2.2.4含量测定方法进样 6 次,以考察进样精密度。每次进样量为 10 μL,测定橙皮苷的峰面积。结果:平均值为539 950.7,RSD=0.4%(n=6)。结果表明:进样精密度良好。
2.2.8 供试液稳定性
取同一供试液,考察 8 h 内供试液的稳定性。分别于0,1,2,4,6,8 h 时进样测定。结果:橙皮苷的峰面积平均值为542 343.4,RSD为 0.7%(n=6)。结果表明:供试液在 8 h 内稳定。
2.2.9 重复性试验
对同一批号的样品,过滤制备 6 份供试品溶液,按2.2.4含量测得法进样测定,各进样10 μL。结果:6 份供试液的橙皮苷成分的含量平均值为 0.04 398 g/L,RSD为 0.6%(n=6)。结果表明:本法的重复性良好。
2.2.10 定量限的测定
取橙皮苷对照品溶液,逐级稀释后进样测定,直观法测得定量限为 11.6ng。
2.2.11 回收率测定
精密量取已知含量的样品 5 mL(含量为 0.043 98 g/L),精密加入橙皮苷对照品溶液 4 mL(质量分数为 0.067g/L),摇匀,即得加样回收供试液,同法进行测定,结果:平均加样回收率为96.50%, RSD=1.28%(n=7)。见表1。
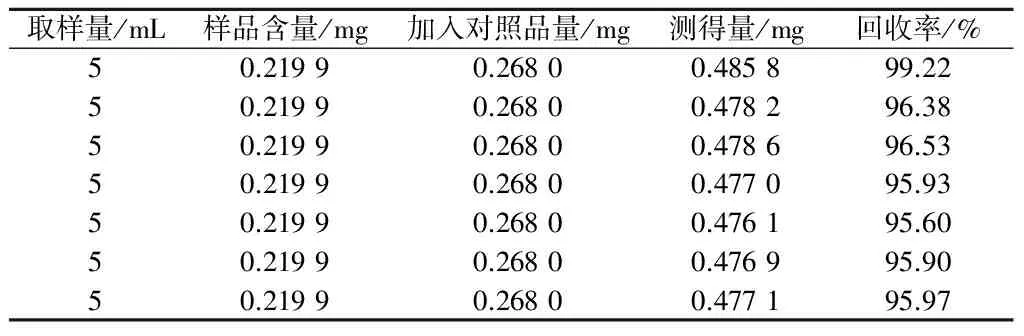
表1 加样回收试验结果
2.2.12 3批样品含量测定
取五加皮酒样品3批(批号依次为20170101,20170102,20170103),按2.2.4含量测得法操作进行测定。结果:3批样品中的橙皮苷的含量依次为0.0439,0.0441和0.0442 g/L。
3 讨 论
本品方中的主要药味为五加皮和陈皮。由于五加皮没有合适的含量测定指标成分,故选取陈皮中的橙皮苷成分为含量测定的指标成分。有关橙皮苷含量测定的研究报道较多。笔者按照《中国药典》中陈皮的含量测定方法,采用高效液相色谱法测定方中橙皮苷成分[2-3]。有文献[4]以高效液相色谱法测定五加皮酒中的橙皮苷和齐墩果酸含量,但其采用的是柱切换技术,方法比较繁琐,流动相比较复杂,测定结果的变异系数较大。笔者的方法较之更为简便准确。当归、川芎和独活中均含有挥发性成分,故本研究采用乙醚萃取制备供试品溶液,参照《中国药典》对这几种药材的薄层色谱鉴别方法进行改进,结果满意。
在含量测定方法研究过程中,笔者还采用过以下几种方法:①取本品,加氯仿振摇脱脂,水层水浴挥去溶剂,再以无水乙醇分次溶解,滤过,制备供试液;②取本品,挥去溶剂,残渣加甲醇分次溶解,滤过,制备供试液;③取本品,挥去溶剂,加入硅藻土,搅拌均匀,挥干溶剂,再加入无水乙醇,水浴回流提取,制备供试液;④取本品,加乙酸乙酯振摇提取,水浴挥去溶剂,残渣分次加40%甲醇使溶解,制备供试液。结果表明,采用前两种方法时,由于本品中含有大量蔗糖,易粘在瓶壁上,影响溶液的转溶;蔗糖沉淀中亦裹挟了较多的成分,影响分析结果。方法③采用硅藻土为吸附剂,目的是为了除去样品中的糖等杂质成分,但高效液相色谱实验结果表明,本法亦造成橙皮苷成分损失。方法④采用乙酸乙酯溶剂萃取样品中的橙皮苷成分,但色谱结果表明,本法提取率较低。故上述方法均不宜采用。本文所采用的供试液制备方法简便易行,准确可靠。
[1]中华人民共和国卫生部药典委员会.中华人民共和国卫生部药品标准:中药成方制剂:第一册[M].北京:中华人民共和国卫生部药典委员会,1989.
[2]国家药典委员会.中华人民共和国药典:四部[M].北京:中国医药科技出版社,2015.
[3]刘杰,任孝德,屠万倩.健儿消食口服液质量标准的改进[J].中医研究,1997年,19(4): 8-9.
[4]杨江丰,沈利君.HPLC法测定五加皮酒中橙皮苷及齐墩果酸的含量[J].中成药,1997,19(4): 8-9.
猜你喜欢
中国测试(2022年4期)2022-05-10
世界科学技术-中医药现代化(2021年9期)2021-12-31
中医眼耳鼻喉杂志(2021年2期)2021-07-21
幸福·健康版(2018年3期)2018-03-23
移动信息(2017年3期)2017-12-28
食品与健康(2017年9期)2017-09-13
中成药(2017年5期)2017-06-13
恋爱婚姻家庭·养生版(2017年2期)2017-02-15
饮食科学(2016年4期)2016-07-06
饮食科学(2015年2期)2015-09-24
