
噬血细胞性淋巴组织细胞增多症的病因和发病机制的研究进展
2018-02-28 07:09:48施清怡肖永红
浙江医学 2018年2期
施清怡 肖永红
噬血细胞性淋巴组织细胞增多症(hemophagocytic lymphohistiocytosis,HLH),又称噬血细胞综合征,不是一个独立的疾病,而是多种致病因素引起的先天性或者获得性免疫系统自我调节作用及抑制作用缺陷导致的高炎症反应性疾病。其病因复杂,发病机制不完全明确。因此,本文对近年来HLH的病因和发病机制的研究进展作一综述。
1 HLH的病因
1.1 原发性HLH 原发性HLH是一种罕见的遗传性疾病,多为常染色体或性染色体隐性遗传,其分型和相关基因突变见表1[1-4]。结合国际免疫学会联盟(IUIS)制定的关于原发性免疫缺陷病分类和诊断的临床指南(2015年版)[3],原发性 HLH可分为两类:(1)家族性HLH(familial HLH,FHL)。根据是否伴随色素减退,FHL可再分为两类:①FHL不伴色素减退:包括FHL-2~FHL-5四个亚型以及EB病毒(EBV)感染相关的X连锁淋巴增殖综合征(X-linked lymph proliferative disease,XLP)。FHL-2~FHL-5所对应的4种突变基因分别为PRF1基因、UNC13D基因、STX11基因和 STXBP2基因。这4种基因都会影响细胞毒性颗粒的胞吐和功能[5]。在FHL病例中,FHL-2约占 20%~40%,FHL-3约占20%~25%,FHL-4约占14%~21%。另外,同一类型FHL的发病率在不同种族之间存在显著差异[1]。FHL-2的突变基因为PRF1基因,编码穿孔素,是重要的细胞溶解蛋白。FHL-3~FHL-5的突变基因分别编码神经突触前膜胞内蛋白13结合蛋白4(Munc13-4)、突触融合蛋白11(syntaxin11)、神经突触前膜胞内蛋白18结合蛋白2(Munc18-2)。上述3种蛋白都参与细胞毒性颗粒(包括穿孔素)的转运和/或细胞膜溶解的过程,因此这些蛋白的缺失都会影响穿孔素的分泌和功能,从而影响细胞毒性细胞的杀伤靶细胞活动[6]。XLP又分为XLP-1和XLP-2,其突变基因分别为SH2D1A和XIAP/BIRC4。XLP-1和XLP-2患者常伴有低丙种球蛋白血症,可引起机体免疫功能下降,易受EBV感染。SH2D1A编码的信号淋巴细胞活化分子相关蛋白(SAP),可调节自然杀伤细胞(NK细胞)和T淋巴细胞的生长,对杀伤淋巴瘤细胞也起一定作用,因此XLP-1可出现NK/T细胞缺乏,细胞毒性反应被削弱,更容易发展为淋巴瘤[7]。另外已发现XLP-1患者体内存在记忆B细胞成熟障碍的现象,从而导致XLP-1患者接种疫苗失效[8]。X连锁凋亡抑制蛋白(XIAP)/BIRC4编码XIAP,相比XLP-1、XLP-2患者更容易患结肠炎等疾病,但鲜有出现淋巴瘤的[1]。②FHL伴色素减退:包括格里塞利综合征2型(Griscelli syndrome typeⅡ,GS-2)、先天性白细胞颗粒异常综合征(Chediak-Higashis yndrome,CHS)、海普综合征 2 型(Hermansky-Pudlak syndrome typeⅡ,HPS-2)、海普综合征 9型(Hermansky-Pudlak syndrome type Ⅸ,HPS-9)。(2)淋巴细胞增生综合征:与EBV感染相关,包括IL-2诱导型T细胞蛋白激酶(ITK)基因、CD27基因、镁离子转运基因(MAGT1)突变分别造成的ITK缺失、CD27缺失、X连锁免疫缺陷病伴镁离子缺陷EBV感染和肿瘤形成。这些基因突变都会呈现不同的免疫缺陷及免疫紊乱表现。ITK缺失的患者常常出现严重的低丙种球蛋白血症以及CD4+T细胞、NK/T细胞急剧减少。当机体感染EBV后,EBV阳性B淋巴细胞大量增殖,导致机体免疫紊乱[9]。CD27是一种淋巴细胞协同刺激分子,CD27缺失会导致T细胞介导的特异性免疫应答受损。

表1 原发性HLH分类和相关的基因突变
1.2 继发性HLH 继发性HLH约占HLH的90%,在各个年龄段均可发病,主要与感染、恶性肿瘤等疾病相关。该病在不同国家的主要病因是不同的,这暗示着该病可能与某一特定的基因背景或者某些可疑的致病因素(尤其是感染)有关[10]。
继发性HLH主要病因包括:(1)感染因素:病毒是最常见的病原体,其中最常见为EBV,其他还包括HIV、疱疹病毒(单纯疱疹病毒、人类疱疹病毒-8、水痘带状疱疹病毒)、巨细胞病毒、肝炎病毒、流感病毒、人细小病毒B19、柯萨奇病毒、登革热病毒、腺病毒Ⅱ型、麻疹病毒等;细菌包括结核分枝杆菌、嗜麦芽寡养单胞菌、布氏杆菌、立克次体菌属、葡萄球菌菌属、大肠杆菌等;寄生虫包括利什曼原虫、疟原虫、弓形虫等;真菌包括组织胞浆菌属等;其余还有支原体、衣原体等。(2)肿瘤:绝大多数为血液系统肿瘤,包括淋巴瘤、白血病、Castleman’s病等,其余还有实体肿瘤。(3)自身免疫系统疾病:系统性红斑狼疮(SLE)、成人Still病、类风湿性关节炎、炎性肠病、系统性硬化病、干燥综合征、多肌炎、原发性胆汁性肝硬化、坏死性淋巴结炎等。巨噬细胞活化综合征(MAS)是风湿性疾病相关的HLH,是风湿性疾病的极其严重的并发症,多见于系统性幼年型特发性关节炎、SLE、成人 Still病[11]。(4)其他事件或疾病:包括移植(肾脏、造血干细胞等)、手术或活检、接种疫苗或急性受伤、血液透析、妊娠等。还有部分继发性HLH的病因并不明确[10,12-14]。
2 HLH的发病机制
2.1 细胞杀伤作用缺陷 当穿孔素/颗粒酶依赖的细胞杀伤机制存在缺陷时,细胞毒性淋巴细胞无法杀伤靶细胞,导致免疫系统自我调节及抑制作用被破坏。
2.1.1 细胞毒性颗粒的胞吐过程 细胞毒性淋巴细胞包括CTL和NK细胞。尽管这两类细胞识别靶细胞的过程存在差异,但是杀伤靶细胞的关键途径是相同的。一旦识别靶细胞,细胞毒性淋巴细胞会通过半胱天冬酶依赖或非依赖途径释放细胞毒性颗粒。细胞毒性颗粒内含具有细胞溶解作用的蛋白质,主要为穿孔素、颗粒酶。穿孔素在胞外Ca2+存在的条件下,构象发生变化,从而结合并插入靶细胞的细胞膜,在靶细胞膜上形成孔隙,因此改变胞内渗透压,细胞膨胀破裂。同时细胞膜上的孔隙,使颗粒酶进入靶细胞,协同杀伤靶细胞[15]。
细胞毒性颗粒的胞吐过程见图1,包括细胞毒性淋巴细胞与靶细胞之间形成免疫突触,随后微管组织中心负责在连接处定位,使包裹有细胞溶解颗粒的囊泡沿着微管蛋白运输,不断锚定、发育成熟并与细胞膜融合,最终细胞溶解颗粒通过胞吐作用分泌至靶细胞。在囊泡转运后期,免疫突触对囊泡的调控是通过形成囊泡-SNARE蛋白(如VAMP)与靶细胞膜-SNARE蛋白(如Syntaxin11或SNAP23/25/29)复合物发挥作用的。但是关于SNARE复合物的明确的生物学功能尚待进一步研究[15]。上述胞吐过程需要几个重要的蛋白质,如Lyst、Ap3β1、Syntaxin11、Rab27a、Munc13-4、Munc18-2 等,其功能与相应疾病见表1。目前除了XLP与一些先天性免疫缺陷疾病外,其他已知的原发性HLH的发病原因都是因为基因突变,编码异常蛋白质,影响细胞毒性颗粒的胞吐和功能[1],导致靶细胞不能得到及时地清除。继发性HLH的发病机制与原发性HLH相似,主要是由于穿孔素/颗粒酶依赖的细胞杀伤机制存在缺陷,免疫系统自我调节及抑制作用被破坏,但是进一步的发病机制尚不清楚。随着基因检测的应用,在许多继发性HLH患者中发现了FHL相关的突变基因的杂合子改变或者多态性现象[1,16]。比如,多项研究发现部分MAS患者中存在UNC13d、PRF1、STX11、STXBP2 和 RAB27a基因中的单个或多个基因突变[17]。这导致原发性与继发性HLH的区分更加困难。
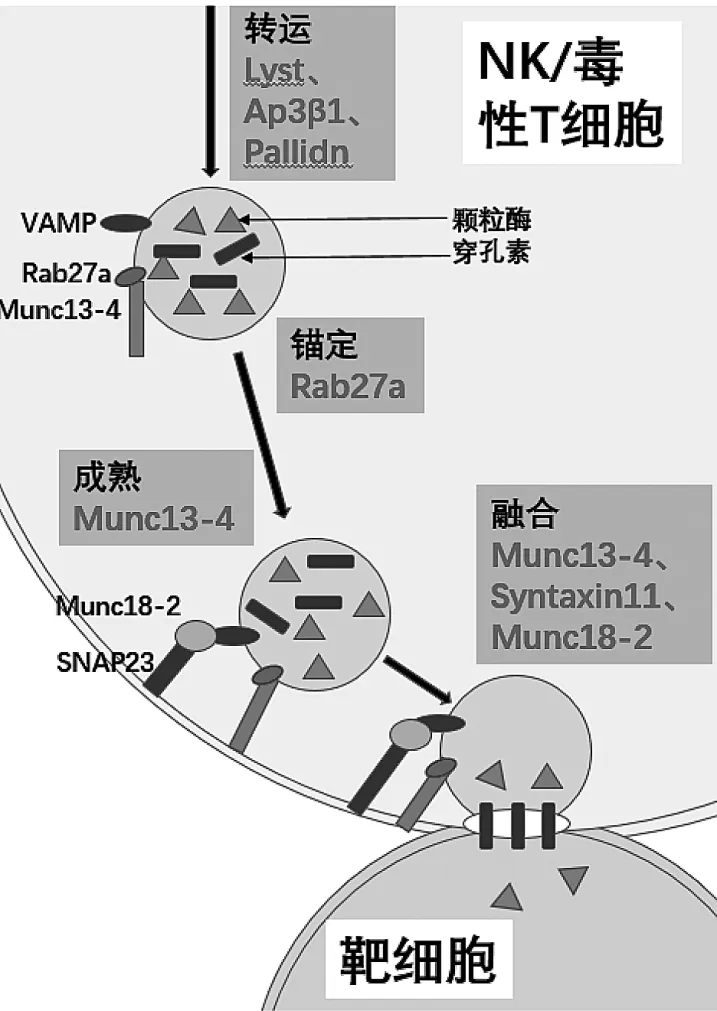
图1 细胞毒性颗粒的胞吐过程
2.1.2 细胞毒性颗粒缺失 现研究发现正常情况下辅助性T细胞亚群1(Th1细胞)通过穿孔素依赖途径清除临界数量的抗原递呈细胞,从而抑制免疫反应。当穿孔素缺失时,该反馈机制也受到影响,导致过度免疫应答[18]。另外,细胞毒性颗粒的缺失,可导致细胞毒性淋巴细胞无法脱离靶细胞,导致细胞毒性细胞持续增生,增生时间可延长至正常的5倍[6]。在这个过程中,Ca2+持续进入细胞毒性细胞,引发大量的促炎性细胞因子和趋化因子的分泌。
2.2 高炎症反应 HLH中活化的T细胞分泌大量细胞因子,其中IFN-γ介导的炎症反应对于HLH的临床表现起重要作用[3]。IFN-γ可导致骨髓细胞过度活化,刺激巨噬细胞分泌过量的细胞因子,如IL-1、IL-4、IL-6、IL-8、IL-10、IL-12、IL-18 及 TNF-α 等,使 Th1/Th2细胞比例失衡。Th1过度活化,又会分泌大量的细胞因子,如IFN-γ、IL-6、TNF-α、粒细胞-巨噬细胞集落刺激因子等细胞因子和趋化因子,导致多系统产生炎性反应,形成了严重的细胞因子风暴[2,19]。另外,FHL的高炎症反应可能也与Toll样受体(TLR)介导的信号传导通路的激活而导致机体固有免疫系统的活化有关[5]。
CTL、巨噬细胞和促炎性细胞因子(如TNF-α)浸润肝、脾、淋巴结、骨髓及中枢神经系统等组织器官,并形成致命性的炎症状态,从而损伤机体组织,出现肝功能异常、肝脾肿大、淋巴结肿大、高铁蛋白血症、中枢神经系统症状等临床表现。例如,活化的T淋巴细胞,分泌大量的可溶性CD25(sIL-2r);组织细胞或巨噬细胞释放大量的铁蛋白,导致高铁蛋白血症;TNF-α和IL-1是常见的趋化因子,趋化中性粒细胞和单核细胞至感染部位;大量的TNF-α、IL-1、IL-6会引起持续性发热,从而造成组织损伤;活化的巨噬细胞在骨髓及其他器官中吞噬血细胞以及造血过程中机体处于高铁蛋白血症状态,均导致血细胞减少症;高水平的TNF-α抑制脂蛋白酶活性,导致高甘油三酯血症;活化的巨噬细胞表达大量的纤维蛋白溶解酶原活化因子,导致低纤维蛋白血症[1,19]。
3 小结
HLH是一种病因复杂,发病机制尚未完全阐明,临床表现缺乏特异性的高炎症反应综合征。病因不同,其治疗方案与预后评估也有差异,因此在HLH诊治过程中应拓宽思路,进行详细的病因筛查。同时深入探究HLH发病机制,进一步提升治疗水平。
[1] Madkaikar M,Shabrish S,Desai M.Current updates on classification,diagnosis and treatment of hemophagocytic lymphohistiocytosis(HLH)[J].The Indian Journal of Pediatrics,2016,83(5):434-443.doi:10.1007/s12098-016-2037-y.
[2] 李佩章,黄玲莎.噬血细胞综合征研究进展[J].中国医学创新,2014,11(24):149-151.doi:10.3969/j.issn.1674-4985.2014.24.051.
[3]Picard C,Al-Herz W,Bousfiha A,et al.Primary immunodeficiency diseases:an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015[J].Journal of Clinical Immunology,2015,35(8):696-726.doi:10.1007/s10875-015-0201-1.
[4] Allen CE,Mcclain KL.Pathophysiology and epidemiology of hemophagocytic lymphohistiocytosis[J].Hematology,2015,2015(1):177-182.doi:10.1182/asheducation-2015.1.177.
[5]JankaGE,Lehmberg K.Hemophagocyticsyndromes--anupdate[J].Blood Reviews,2014,28(4):135-142.doi:10.1016/j.blre.2014.03.002.
[6] Voskoboinik I,Whisstock JC,Trapani JA.Perforin and granzymes:function,dysfunction and human pathology[J].Nature Reviews Immunology,2015,15(6):388-400.doi:10.1038/nri3839.
[7]Weng X,Liao CM,Bagchi S,et al.The adaptor protein SAP regulates type II NKT-cell development,cytokine production,and cytotoxicity against lymphoma[J].European Journal of Immunology,2014,44(12):3646-3657.doi:10.1002/eji.201444848.
[8]Nichols KE,Ma CS,Cannons JL,et al.Molecular and cellular pathogenesis of X-linked lymphoproliferative disease[J].Immunological Reviews,2005,203:180-199.doi:10.1111/j.0105-2896.2005.00230.x.
[9] Ghosh S,Bienemann K,Boztug K,et al.Interleukin-2-inducible T-cell kinase(ITK)deficiency-clinical and molecular aspects[J].J Clin Immunol,2014,34(8):892-899.doi:10.1007/s10875-014-0110-8.
[10] Ramoscasals M,Britozerón P,Lópezguillermo A,et al.Adult haemophagocytic syndrome[J].Lancet,2014,383(9927):1503-1516.doi:10.1016/S0140-6736(13)61048-X.
[11] Janka GE.Familial and acquired hemophagocytic lymphohistiocytosis[J].Annual Review of Medicine,2012,63(1):233-246.doi:10.1146/annurev-med-041610-134208.
[12] 丁思睿赟,郭鹏翔.噬血细胞综合征的病因及诊断进展[J].中外医疗,2015,10:197-198.doi:10.3969/j.issn.1674-0742.2015.10.093.
[13] 李硕,王晶石,王旖旎,等.147例噬血细胞综合征病因及预后分析[J].临床血液学杂志,2014,27(4):559-563.doi:10.13201/j.issn.1004-2806.2014.07.005.
[14] George MR.Hemophagocytic lymphohistiocytosis:review of etiologies and management[J].Journal of Blood Medicine,2014,5:69-86.doi:10.2147/jbm.s46255.
[15] Ishii E.Hemophagocytic lymphohistiocytosis in children:pathogenesis and treatment[J].Frontiers in Pediatrics,2016,4:47.doi:10.3389/fped.2016.00047.
[16]Gupta S,Weitzman S.Primary and secondary hemophagocytic lymphohistiocytosis:clinical features,pathogenesis and therapy[J].Expert Review of Clinical Immunology,2010,6(1):137-154.doi:10.1586/eci.09.58.
[17] Bracaglia C,Prencipe G,De Benedetti F.Macrophage activation syndrome:different mechanisms leading to a one clinical syndrome[J].PediatricRheumatologyOnlineJournal,2017,15(1):5.doi:10.1186/s12969-016-0130-4.
[18] Terrell CE,Jordan MB.Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+)T cells and dendritic cells[J].Blood,2013,121(26):5184-5191.doi:10.1038/nri3839.
[19] 张滔,陈建行,熊梅.噬血细胞综合征研究进展[J].临床和实验医学杂志,2013,23:1938-1941.
猜你喜欢
新传奇(2022年51期)2023-01-04 21:51:14
分子诊断与治疗杂志(2022年11期)2022-12-23 13:13:18
基层中医药(2020年2期)2020-07-27 02:45:56
小哥白尼(野生动物)(2019年5期)2019-08-27 00:53:38
中医眼耳鼻喉杂志(2019年3期)2019-04-13 05:26:50
中国免疫学杂志(2019年3期)2019-03-13 02:11:14
神州·中旬刊(2019年1期)2019-02-12 08:47:44
小布老虎(2017年1期)2017-07-18 10:57:27
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:10
发明与创新(2015年33期)2015-02-27 10:40:02
