
常染色体显性多囊肾病的预后评估及治疗
2018-02-02 08:03周晨辰梅长林
协和医学杂志 2018年1期
薛 澄,周晨辰,梅长林
第二军医大学上海长征医院肾内科 解放军肾脏病研究所, 上海 200003
常染色体显性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)是最常见的遗传性肾病,患病率为1‰~2‰[1],在欧洲、美国、日本均属于罕见病[2]。ADPKD是引起终末期肾病(end-stage renal disease,ESRD)的第4位病因,约占西方透析人数的10%。ADPKD患者进入ESRD的中位年龄为58岁,主要依靠肾脏替代治疗(renal replacement therapy,RRT)维持生命[3]。我国约有150万例ADPKD患者,给社会带来了沉重的经济负担[2]。多年来,ADPKD一直被认为是一种无法治疗的疾病。自2014年起,情况略有改观,经多国监管部门批准,托伐普坦成为延缓ADPKD进展的第一种有效药物。本文主要综述ADPKD的临床特征、预后评估及治疗等方面的进展。
1 临床特征
ADPKD主要表现为双侧肾囊肿且逐渐发展,肾脏体积进行性增大,肾小球滤过率(glomerular filtra-tion rate,GFR)逐步降低,常出现高血压、腰痛、血尿、囊肿或尿路感染等并发症[2]。ADPKD属多系统性疾病,35岁以上患者中超过90%合并肝囊肿、胰腺囊肿、颅内动脉瘤、结肠憩室或心脏瓣膜异常[4]。其疾病特征和进展在家族内及家族间有很大变异性,约半数患者可因临床症状不明显而未被诊断为ADPKD[5]。根据欧洲肾脏协会-欧洲透析和移植协会(European Renal Association-European Dialysic and Transplant Association,ERA-EDTA)发布的数据,确诊的患病率仅为0.329‰[6]。多数ADPKD患者在很长一段时间内病程进展缓慢,尽管数以千计的微小囊肿已经形成,但未发生囊性病变的肾单位可在长时间内代偿,维持GFR相对稳定。直至在疾病晚期时,由于增大增多的囊肿对残余肾单位的压迫和肾间质炎症和纤维化的改变,使患者进入肾衰竭的速度显著加快。研究发现,肾脏大小与GFR成反比,同样肾脏体积的患者中男性较女性肾功能损伤程度更重[3]。此外,实验室指标中的肽素、中性粒细胞明胶酶蛋白、单核细胞趋化因子- 1、尿酸等可能是预测ADPKD进展的敏感指标[7]。
2 致病基因
PKD1和PKD2基因突变约占已发现ADPKD患者的90%,分别编码多囊蛋白1和2,未发现突变者约占8%~10%[4]。两种多囊蛋白可形成异二聚体,其功能为非选择性钙离子通道[8]。近期,GANAB被证实为ADPKD第3种致病基因,但只占PKD1和PKD2突变阴性患者的一小部分[9]。位于染色体16p13.3的PKD1突变约占81%,位于染色体4q21- 22的PKD2突变约占10.5%~22%[10]。ADPKD自发突变率高达8%。而疾病进展快慢则可部分归因于特异性基因突变。相比PKD1,PKD2突变患者疾病进程更为缓慢,进入ESRD的中位年龄约晚20~25岁[11]。
3 病理生理
PKD1和PKD2等位基因在感染、毒素和环境的作用下,易发生“二次打击”,产生突变,使多囊蛋白失去功能,引起肾小管细胞周期调控和代谢异常,上皮细胞增殖,形成微小囊肿,阻塞肾小管,使液体聚积。同时,多囊蛋白复合体结构和功能的异常可引起钙离子内流信号减弱,导致肾小管细胞表面纤毛细胞极性和迁移的改变,使Na+-K+-ATP酶异位于肾小管细胞腔内膜,向囊腔分泌液体,促进肾囊肿的增大[8]。血管加压素(arginine vasopressin,AVP)和环磷酸腺苷(cyclic adenosine monophosphate,cAMP)相关信号通路在ADPKD囊肿发展过程中发挥了重要的作用。哺乳动物雷帕霉素靶蛋白复合物1(mammalian target of rapamycin complex 1,mTORC1)信号通路在ADPKD中异常活化,促进cAMP表达增加[12]。cAMP的增加导致囊肿衬里上皮细胞中离子和水转运失调引发囊肿,进而不断进展[12]。AVP的V2受体拮抗剂可有效降低AVP促进的细胞内源性cAMP的水平。目前纳入ADPKD临床研究的哺乳动物雷帕霉素靶蛋白通路抑制剂西罗莫司、依维莫司,及V2受体拮抗剂托伐普坦主要针对以上病理生理异常。生长抑素类似物则主要通过上调Gα受体降低胞内cAMP水平。
4 影像学及基因诊断
美国多囊肾病影像研究协会(Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease, CRISP)提出肾脏增大是ADPKD首先出现的临床表现[13],典型表现为双肾分布多个大小不一囊肿,在肾功能开始下降前患者总肾脏体积(total kidney volume,TKV)>1 L[14]。因此超声检查是目前诊断ADPKD的主要方法,其准确性高达96%[15],主要依据包括家族史、肾囊肿数量及发病年龄。根据Pei[16]等最新的超声诊断标准:15~39岁患者,单侧或双侧囊肿数目>3;40~59岁每侧肾脏囊肿数目>2;>60岁患者每侧肾脏囊肿数>4,且家族史阳性的患者可诊断为ADPKD。
基因诊断可以检测出约90%ADPKD患者的突变基因,剩余10%符合ADPKD诊断的患者不存在PKD1和PKD2突变。基因检测早期使用聚合酶链式反应(polymerase chain reaction,PCR)技术和Sanger测序,近年来随着人类基因组计划的全面实施完成,二代测序技术(next generation sequencing, NGS)已成为目前主要的基因检测方法。相比PCR和Sanger,NGS测序速度更快,成本更低,但敏感性和准确性不及Sanger测序。基因诊断的临床指征包括:(1)排除肾脏捐献者是否为非典型ADPKD患者;(2)家族史阴性患者需明确诊断以排除其他囊肿性疾病;(3)胚胎植入前需行遗传学诊断(preimplantation genetic diagnosis,PGD);(4)需明确基因型,以对患者预后或疾病进展速度作出评估[17]。由于费用昂贵(约¥4000~9000/次),技术复杂,目前基因检测尚未常规用于ADPKD诊断。
上海长征医院通过PGD技术成功阻断ADPKD遗传,自38个体外受精胚胎中通过多次退火环状循环扩增技术(multiple annealing and looping-based amplifica-tion cycles,MALBAC)[18]筛选出6个不携带致病突变、无染色体异常的胚胎,其中1例已成功移植回母体存活发育,在孕18周的羊水穿刺检查中未检出携带致病基因突变,近期诞生了国内第1例父亲为ADPKD患者的健康新生儿,关于此技术应用的多中心临床研究正在进行中。对家族史阳性儿童进行基因检测的实际意义有限,主要由于缺乏可以控制患儿疾病进展的药物,通过监测血压可以监控疾病进展。
5 预后评估
ADPKD患病个体之间肾病进展差异很大,即便同一家族,患者的发病年龄和进入ESRD速度也存在较大差异,因此对患者预后进行预测具有重要临床意义。预后评估模型可有效分析ADPKD患者预后风险,协助临床医生制定决策。预测患者预后与多种影响因素密切相关,其中TKV是ADPKD肾脏进展的最佳预测因子。改善全球肾脏病预后组织(Kidney Disease: Improving Global Outcomes, KDIGO)2014会议指出磁共振成像(magnetic resonance imaging,MRI)检测患者基线TKV与肾功能衰竭风险密切相关[19]。其他已证实的影响预后因素还包括年龄、性别、肾功能、肉眼血尿及高血压等[20]。
近年来已发表两种重要的预后风险评估模型[21- 22]:梅奥风险评估模型和PROPKD(predicting renal outcome in polycystic kidney disease)评分。美国梅奥风险评估模型主要依据身高校正TKV(表1)[22],而欧洲PROPKD评分主要依据基因型和临床症状出现时的年龄[21]。梅奥模型通过对590例ADPKD患者进行回顾性队列研究,先将患者分为典型(1类)和非典型(2类)两类;进一步根据测定的TKV增长率快慢(MRI或CT检测)将1类患者分为1A至1E五个亚类,1A至1E组的肾小球滤过率估计值(estimated glomerular filtration rate,eGFR)下降速率逐步加快。再通过内部验证(n=162)和外部CRISP队列验证(n=173)发现,1C~1E亚类患者相比1A和1B类患者eGFR年下降值更高[>3 ml/(min·1.73 m2)]。
PROPKD研究基于Genkyst队列研究(1341例ADPKD患者),综合了基因型、性别、出现尿路症状或高血压的年龄(以35岁为界值)等危险因素。研究者根据进展至ESRD的风险将患者分为低、中、高3组。英国肾脏协会根据预后模型和英国国家卫生保健优化研究所(National Institute for Health and Care Excellence,NICE)指南制定的ADPKD诊疗决策流程图见图1。
梅奥模型的优势是根据单个TKV测量指标可预测患者预后风险[22]。但梅奥研究的人群初始eGFR[中位数75 ml/(min·1.73 m2)]较高,且ESRD发病率较低(22%),因而更适合早期PKD患者的预后评估[22]。PROPKD研究中ESRD患者占44.6%,平均年龄高于梅奥研究(54.7岁比44岁),因而PROPKD评分不适用于35岁以下且无症状的ADPKD患者[21]。
ADPKD:常染色体显性多囊肾病;TKV:总肾脏体积;eGFR:肾小球滤过率估计值;ESRD:终末期肾病
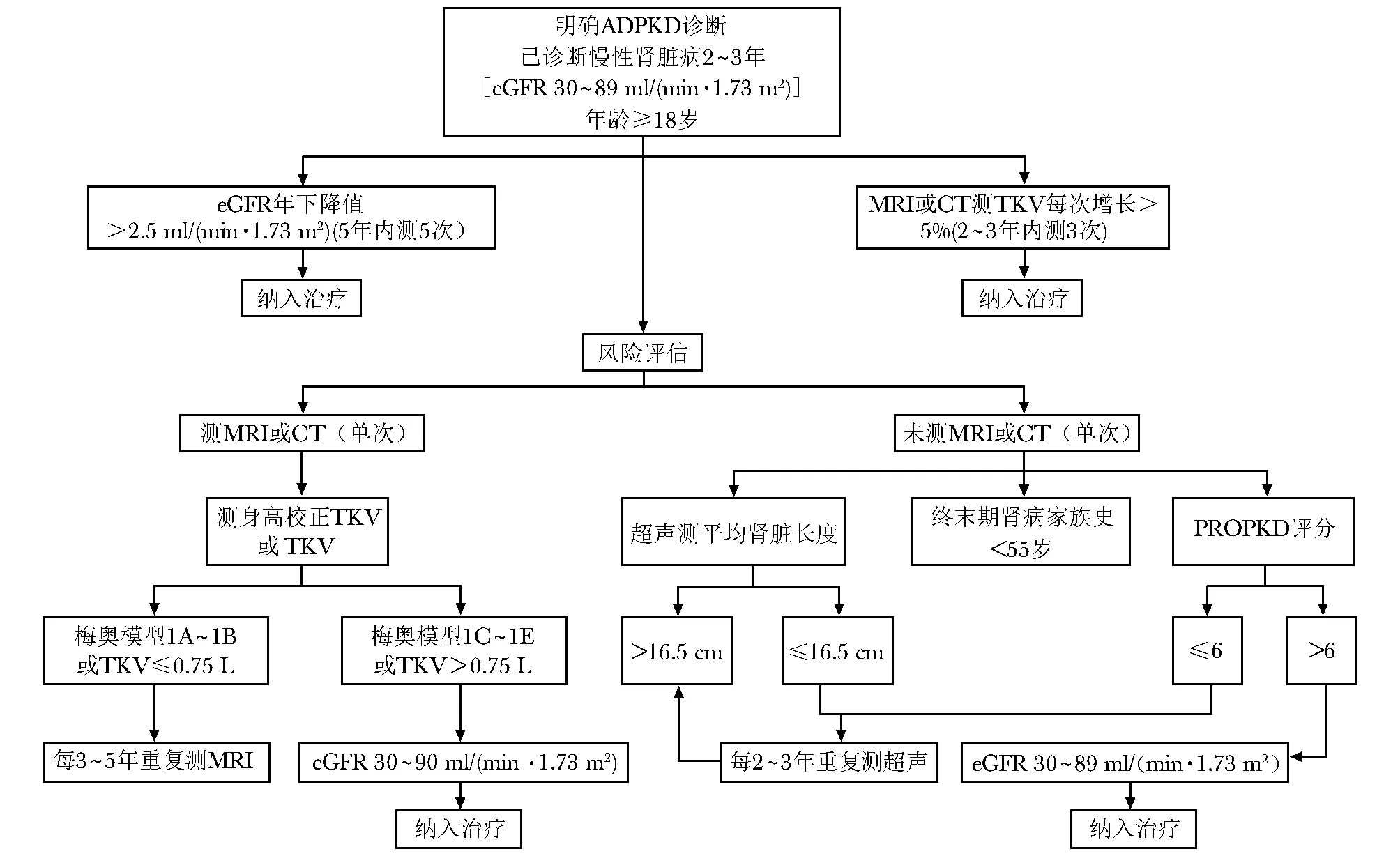
图1NICE指南制定的ADPKD诊疗决策流程图
ADPKD、TKV、eGFR、ESRD:同表1; MRI:磁共振成像; NICE:英国国家卫生保健优化研究所
此外,PROPKD评分的各组内变异可能会影响预测的准确性[19]。因此,ADPKD的预后评估仍需更多大规模的研究验证。模型中加入PKD1截断突变、PKD2、主要差异基因指标、家族史等影响因素可以提高预测效能。
6 治疗进展
长期以来,ADPKD主要依靠支持治疗控制疾病进展,KDIGO 2014会议建议使用支持治疗以减轻ADPKD临床症状,并减少并发症发生率及死亡率[19]。治疗措施包括:低盐饮食、他汀类药物及降压药的使用、每日足量液体摄入(2~3 L/d)、禁烟、避免使用肾毒性药物和摄入咖啡因。
60%~80%的ADPKD患者在疾病早期便出现高血压。肾素血管紧张素系统(renin-angiotensin-aldosterone system,RAAS)异常激活在ADPKD患者并发高血压中发挥重要作用。RAAS拮抗剂已证实为治疗ADPKD高血压的首选药物;血管紧张素转换酶抑制剂(angiotensin-converting enzyme inhibitors,ACEI)相比钙离子拮抗剂可以更好地降低血压、蛋白尿并保护肾功能;ACEI,血管紧张素受体阻滞剂(angiotensin receptor blocker,ARB)和β受体阻滞剂之间在血压和肾脏终点方面无统计学差异[23]。此外,RAAS拮抗剂联合使用(ACEI联合ARB)并无额外肾功能和TKV受益。降压靶目标建议在140/90 mm Hg(1 mm Hg=0.133 kPa)。然而HALT-PKD研究发现,对于快速进展的ADPKD患者,降压靶目标可设定为110/70 mm Hg[24]。对慢性肾脏病(chronic kidney disease,CKD)4期及以上或不能耐受RAAS拮抗剂的患者,可改用β受体阻滞剂[23]。
近年来,临床研究主要集中于特异性抑制囊肿生长的药物,包括mTOR抑制剂、生长抑素类似物、V2受体拮抗剂。mTOR抑制剂的两项大型随机对照研究发现:mTOR抑制剂可有效减缓患者TKV增长,依维莫司组第1年TKV平均增长小于安慰剂组(102 ml比157 ml,P=0.02)[25],但对肾功能的保护作用却不显著[25- 26]。生长抑素类似物奥曲肽的ALADIN研究发现,随访第1年时奥曲肽可有效阻止TKV增长,但在随访第3年,该阻止效应失去了统计学意义(P=0.25)[27]。
目前唯一获批用于ADPKD患者临床使用的是V2受体拮抗剂托伐普坦。托伐普坦已在欧盟、英国、日本、加拿大和韩国获批使用,主要用于控制高风险ADPKD患者的肾病进展。TEMPO 3/4临床试验[28]共纳入1445例ADPKD患者,随机分为晨90 mg/晚30 mg托伐普坦组和安慰剂组,随访3年,建议患者增加每日饮水量。纳入患者eGFR>60 ml/(min·1.73 m2)且TKV>0.75 L。结果发现托伐普坦可显著减慢ADPKD患者TKV的年增长率(2.8%比5.5%,P<0.001),并降低eGFR下降速率。托伐普坦组患者发生eGFR下降25%事件的风险降低了61%,透析前平均肾脏存活时间从4年延长至6.5年。近期发表的TEMPO 4/4试验[29]进一步阐明了长期及早期使用伐普坦的有效性和必要性,随访2年后早用组患者的eGFR下降较晚3年使用托伐普坦的患者慢(P<0.001),两组TKV增长无显著差异。
各个国家使用托伐普坦治疗ADPKD的适应症有所不同[6]。欧洲药物监督管理局发布的托伐普坦适用指征为:可用于CKD 1~3期具有快速进展风险的成年ADPKD患者,用于减慢囊肿进展和肾功能受损[6]。NICE指南不推荐CKD 1期ADPKD患者使用托伐普坦,主要依据来自药物效益成本分析:托伐普坦减缓CKD 1~3期患者eGFR下降速率分别为16%、29%、31%[30]。另一方面,使用托伐普坦要考虑风险和获益平衡,使用药物5年内,每月需监测肝功能,注意口渴、多尿的副作用[30],超过5年的远期并发症目前仍无报道可参考。
7 结语
随着基因技术的发展和成本下降,未来将逐渐普及ADPKD患者的基因诊断;PGD技术可有效阻断ADPKD的遗传,将极大降低ADPKD患儿出生率,提高人口素质,具有重要的临床意义;梅奥风险评估模型和PROPKD评分是较好的预测模型,已成为临床医生决策的重要依据;托伐普坦是ADPKD的首个针对囊肿生长的特异治疗药物,药物的长期安全性及ADPKD获益人群的细化仍需进一步研究。
[1] Neumann HP,Jilg C,Bacher J, et al. Epidemiology of autosomal-dominant polycystic kidney disease: an in-depth clinical study for south-western Germany [J]. Nephrol Dial Transplant, 2013, 28: 1472- 1487.
[2] Xue C,Zhou CC,Wu M, et al. The clinical manifestation and management of autosomal dominant polycystic kidney disease in China [J]. Kidney Dis(Basel), 2016, 2: 111- 119.
[3] Ong AC,Devuyst O,Knebelmann B, et al. Autosomal dominant polycystic kidney disease: the changing face of clinical management [J]. Lancet, 2015, 385: 1993- 2002.
[4] Sommerer C,Zeier M. Clinical manifestation and manage-ment of ADPKD in western countries [J]. Kidney Dis(Basel), 2016, 2: 120- 127.
[5] Steinman TI. Polycystic kidney disease: a 2011 update [J]. Curr Opin Nephrol Hypertens, 2012, 21: 189- 194.
[6] Mao Z,Chong J,Ong AC. Autosomal dominant polycystic kidney disease: recent advances in clinical management [J]. F1000Res, 2016, 5: 2029.
[7] Chen L,Zhou X,Fan LX, et al. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease[J]. J Clin Invest, 2015, 125: 2399- 2412.
[8] Ong AC,Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex [J]. Kidney Int, 2005, 67: 1234- 1247.
[9] Porath B,Gainullin VG,Cornec-Le Gall E, et al. Mutations in GANAB, encoding the glucosidase Ⅱα subunit, cause autosomal-dominant polycystic kidney and liver disease [J]. Am J Hum Genet, 2016, 98: 1193- 1207.
[10] Audrezet MP,Cornec-Le Gall E,Chen JM, et al. Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients [J]. Hum Mutat, 2012, 33: 1239- 1250.
[11] Ong AC,Harris PC. A polycystin-centric view of cyst formation and disease: the polycystins revisited [J]. Kidney Int, 2015, 88: 699- 710.
[12] Harris PC,Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease [J]. J Clin Invest, 2014, 124: 2315- 2324.
[13] Grantham JJ,Torres VE,Chapman AB, et al. Volume progression in polycystic kidney disease [J]. N Engl J Med, 2006, 354: 2122- 2130.
[14] Chapman AB,Bost JE,Torres VE, et al. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease [J]. Clin J Am Soc Nephrol, 2012, 7: 479- 486.
[15] Bhutani H,Smith V,Rahbari-Oskoui F, et al. A compar-ison of ultrasound and magnetic resonance imaging shows that kidney length predicts chronic kidney disease in autosomal dominant polycystic kidney disease [J]. Kidney Int, 2015, 88: 146- 151.
[16] Pei Y,Obaji J,Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD [J]. J Am Soc Nephrol, 2009, 20: 205- 212.
[17] Simms RJ,Travis DL,Durkie M, et al. Genetic testing in the assessment of living related kidney donors at risk of autosomal dominant polycystic kidney disease [J]. Transplanta-tion, 2015, 99: 1023- 1029.
[18] Zong C,Lu S,Chapman AR, et al. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell[J]. Science, 2012, 338: 1622- 1626.
[19] Chapman AB,Devuyst O,Eckardt KU, et al. Autosomal-dominant polycystic kidney disease(ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes(KDIGO) Controversies Conference [J]. Kidney Int, 2015, 88: 17-27.
[20] Schrier RW,Brosnahan G,Cadnapaphornchai MA, et al. Predictors of autosomal dominant polycystic kidney disease progression [J]. J Am Soc Nephrol, 2014, 25: 2399- 2418.
[21] Cornec-Le Gall E,Audrezet MP,Rousseau A, et al. The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease [J]. J Am Soc Nephrol, 2016, 27: 942- 951.
[22] Irazabal MV,Rangel LJ,Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials [J]. J Am Soc Nephrol, 2015, 26: 160- 172.
[23] Xue C,Zhou C,Dai B, et al. Antihypertensive treatments in adult autosomal dominant polycystic kidney disease: network meta-analysis of the randomized controlled trials [J]. Oncotarget, 2015, 6: 42515- 42529.
[24] Torres VE,Abebe KZ,Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease [J]. N Engl J Med, 2014, 371: 2267- 2276.
[25] Walz G,Budde K,Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease[J]. N Engl J Med, 2010, 363: 830- 840.
[26] Serra AL,Poster D,Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease [J]. N Engl J Med, 2010, 363: 820- 829.
[27] Caroli A,Perico N,Perna A, et al. Effect of longacting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease(ALADIN): a randomised, placebo-controlled, multicentre trial [J]. Lancet, 2013, 382: 1485- 1495.
[28] Torres VE,Chapman AB,Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease [J]. N Engl J Med, 2012, 367: 2407- 2418.
[29] Torres VE,Chapman AB,Devuyst O, et al. Multicenter, open-label, extension trial to evaluate the long-term efficacy and safety of early versus delayed treatment with tolvaptan in autosomal dominant polycystic kidney disease: the TEMPO 4:4 Trial [J]. Nephrol Dial Transplant, 2017. doi: 10.1093/ndt/gfx043. [Epub ahead of print].
[30] Torres VE,Higashihara E,Devuyst O, et al. Effect of tolvaptan in autosomal dominant polycystic kidney disease by CKD stage: results from the TEMPO 3:4 trial [J]. Clin J Am Soc Nephrol, 2016, 11: 803- 811.
猜你喜欢
快乐语文(2019年30期)2020-01-04
快乐语文(2019年36期)2019-01-11
中国医学人文(2018年1期)2018-03-01
心脑血管病防治(2016年6期)2017-01-16
健康管理(2016年9期)2016-10-24
中华老年多器官疾病杂志(2016年7期)2016-04-28
中国实用医药(2016年5期)2016-02-20
中国社区医师(2015年14期)2015-12-24
医学研究杂志(2015年12期)2015-06-10
中国当代医药(2015年22期)2015-03-01
