
中国罕见病研究的现状与未来
2018-02-02 08:03:40徐昊鹏弓孟春张抒扬
协和医学杂志 2018年1期
徐昊鹏,朱 翀,弓孟春,张抒扬
1中国医学科学院 北京协和医学院 临床医学专业,北京 100005 2中国国家罕见病注册系统,北京 100730 3神州数码医疗科技股份有限公司,北京 100080 4中国医学科学院罕见病研究中心,北京 100005 5中国医学科学院 北京协和医学院 北京协和医院,北京 100730
罕见病是一组发病率和患病率极低的疾病,各国家或地区对其定义存在差异,欧盟将罕见病定义为患病率低于1/2000的疾病[1],而美国将罕见病定义为患者人数少于20万人的疾病[2]。一直以来,罕见病给人类社会发展带来了沉重负担,也是现代医疗亟需攻破的重大难题之一。一方面罕见病种类繁多,目前全球估计有7000余种[3],无论是研究力量还是社会关注均较为分散;另一方面罕见病单种疾病患者人数稀少,针对单种疾病开展的研究十分有限,导致人类缺乏对许多疾病发病机制的深入了解;此外,虽然80%以上罕见病具有孟德尔遗传特点[3],但仍存在表型、基因型及基因型-表型分析的技术挑战,导致临床作出准确诊断和治疗均十分困难。
1 全球罕见病研究概况
全球多个国家、多方机构已采取措施,将针对罕见病的研究、诊治及社会关怀作为重要战略部署。1983年美国颁布了《孤儿药法案》[2],随后欧盟、澳大利亚、日本等国家相继出台了罕见病领域的法案[1, 4- 5],推动了罕见病研究与药物研发的进程。包括中国在内的多个国家均开展了单病种或多病种的罕见病注册登记研究[6],这类研究为罕见病的流行病学、发病机制、自然病程和药物研发打下了一定基础。遗传学、生物信息学等学科的发展也对罕见病的研究和诊治起到了极大推动作用。过去30年间,大约3500种单基因遗传性罕见病相关基因已得到明确[3],二代测序技术(next-generation sequencing, NGS)等的快速发展,加速了致病基因的定位。2015年,全外显子组测序技术发现了90%以上的新致病基因[7],几乎全部单基因遗传性罕见病的致病基因将在2020年前得到定位[3]。罕见病治疗药物研发成为罕见病与常见病药物研发的重要组成,2016年美国食品药品监督管理局(Food and Drug Administration,FDA)约41%的获批新药为罕见病治疗药物[8]。同时,孤儿药市场也快速增长,市场份额快速提升[9]。罕见病研究与诊疗的巨大社会、科学价值及效益得以体现。
2 中国罕见病研究现状
中国是一个人口大国,保守估计罕见病患者数目巨大,如何帮助患者解除疾病痛苦、改善生活质量,并借助丰富的病例资源推进罕见病的深入研究,均是需要解决的重大问题。
在罕见病研究领域,多个学会组织、医疗机构及社会公益组织开展了罕见病的研究、诊疗及社会干预工作。2010年起,山东、上海、北京、广东和浙江等省市先后成立了罕见病学会或防治协会,搭建罕见病研究与诊治的协作平台。与此同时,针对罕见病的研究在国内多个中心分别展开,研究涵盖多种疾病基础及临床多个环节。在多种罕见病遗传机制方面,我国研究者报道了多项重要的致病基因及表型-基因型研究成果,如在先天性脊柱侧凸患者中报道了TBX6基因无义突变及一个亚效等位基因的复合遗传模式[10];在高磷血症性家族性肿瘤样钙沉着症(hyperphosphatemic familial tumoral calcinosis, HFTC)患者中报道了两例位于同一家系的GALNT3基因新发突变,为HFTC的表型-基因型关联分析、产前诊断和遗传咨询提供了新的依据[11];在限制性心肌病患者中通过二代测序方法证实了与扩张性心肌病、肥厚型心肌病等心肌病变相关的MYBPC3基因突变与家族性和散发限制性心肌病表型存在致病关联,提示通过二代测序、选择特定的基因panel有助于明确遗传性或散发性限制性心肌病诊断[12]。在罕见疾病的发病机制研究中,我国研究者进行了大量长期的观察研究,如在遗传性肾炎(Alport综合征)患者中发现随着患者年龄增长,肾小球足细胞失去附着、足细胞数量减少,与蛋白尿、肾小球硬化、肾功能减退等临床表现相关[13]。在诊断评估方法方面,我国研究者对常见疾病大类下的罕见病亚型进行研究,如对未接受药物治疗的早发型帕金森病和迟发型帕金森病早期患者的生理功能连接特点进行定位和评估,发现不同疾病亚型的生理功能连接存在差异,此类研究的深入开展将进一步揭示帕金森病的发病机制[14]。此外,我国临床研究者在某些罕见病如肺动脉高压[15]、淋巴管平滑肌瘤病[16]等的临床诊断和治疗方案优化方面取得了一定研究成就,得到了国际学术界的认可。
以中国罕见病发展中心为代表的一系列罕见病患者组织的成立,为罕见病患者提供了多维度社会支持,并推动了罕见病立法。例如中国罕见病发展中心发布《中国罕见病参考名录》,并启动中国罕见病患者登记项目,可系统地了解罕见病患者生存现状,并为科学研究、政策保障提供支持与参考。患者组织还通过鼓励患者参与、联络患者群体及搭建信息平台等方式支持罕见病注册研究。目前,罕见病患者组织已成为罕见病研究、诊疗与社会参与体系中的重要组成部分。
2017年10月9日中共中央办公厅、国务院办公厅印发了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》[17],提出支持罕见病治疗药品、医疗器械研发,包括允许罕见病治疗药品、医疗器械注册申请人提出减免临床试验的申请,对境外已批准上市的罕见病治疗药品、医疗器械,可附带条件批准上市;中国国家卫生计生委罕见病诊疗与保障专家委员会在北京协和医院设立了委员会办公室,负责委员会日常工作;科技部“十三五”国家重点研发计划“精准医学研究重点专项”启动了“罕见病临床队列研究”与“中国人群重要罕见病的精准诊疗技术与临床规范研究”等课题,以此为开端,中国启动了首个全国性罕见病注册登记研究。作为全国疑难重症诊治中心,北京协和医院牵头此项罕见病临床队列研究,以中国国家罕见病注册系统(National Rare Diseases Registry System, NRDRS)为平台,联合20家国内顶尖教学医院,登记研究50余种至少5万例相关罕见病例,建设多组学数据库与多中心临床生物样本库。根据既定的路线图,NRDRS将为罕见病精准分型、诊断、治疗和预防提供决策依据(图1)[18]。由于研究病种繁多、研究人群庞大,临床表型与基因型数据的采集、挖掘与分析能力对罕见病临床队列研究而言尤为关键。针对表型数据分析,NRDRS致力于建设符合国际主流标准的信息系统,目前已部分对接中文人类表型本体(Chinese human phenotype ontology,CHPO)及中文观测指标标识符逻辑命名与编码系统(Logistic observation identifiers names and codes,LOINC),并持续推进其他中文术语体系和本体的开发。在基因型数据采集领域,采用先进的高通量测序技术及信息处理分析技术,实现基因型数据的准确采集及有效解读,为研究与诊治提供保障。此外,NRDRS还将发挥平台优势,建设并完善数据共享机制,整合各类临床及知识资源,并推动多个利益相关方合作,共同推进罕见病研究与诊治。
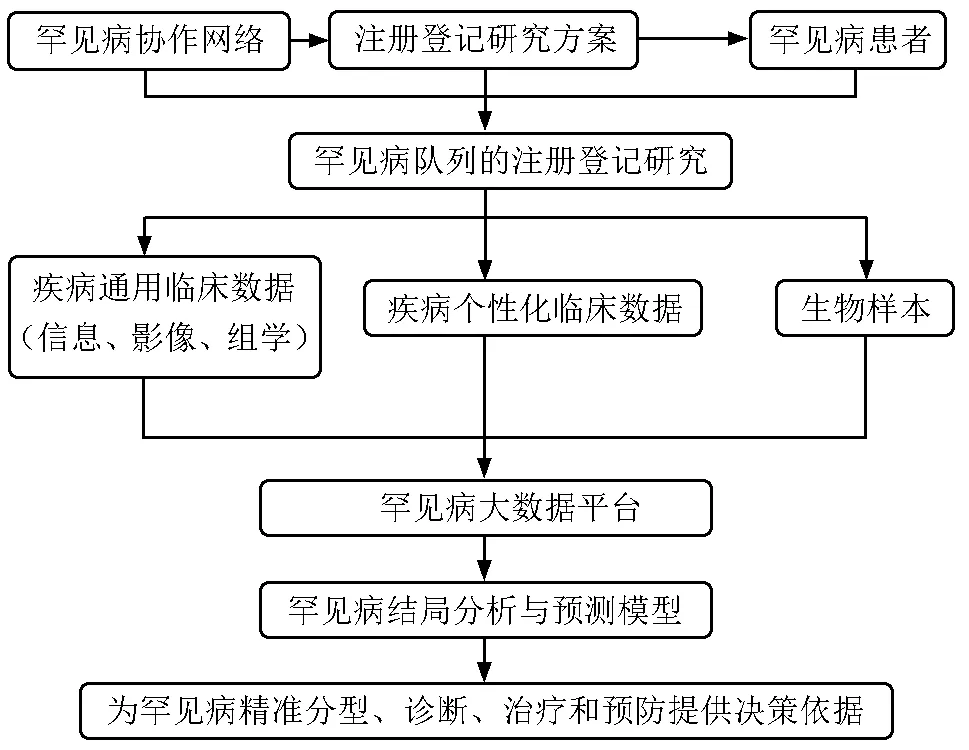
图 1 中国国家罕见病注册系统路线图
同时,中国在罕见病领域依然面临着重大挑战。首先,目前国内对罕见病暂无明确定义,对罕见病干预与社会保障政策实施仍存在一定困难。其次,罕见病研究力量分散于不同地区、缺乏有效的资源统筹与协调机制,致使罕见病整体数据不足、研究样本量小、数据质量不高、长期随访难。再次,在罕见病临床诊疗、遗传咨询、药物研发和生产领域,存在专业人才不足、临床诊疗路径不完整的问题,进而导致患者转诊次数多、误诊及漏诊率高、有效干预措施缺乏,最终致使医疗资源浪费。最后,国内罕见病患者组织的力量和公众认知程度明显落后于发达国家,如美国罕见病协会纳入了250余家患者组织,而截至2016年底,中国仅注册70余家罕见病患者组织[19]。在这样的形势下,协调各方力量、构建罕见病研究诊疗体系、呼吁社会广泛参与是中国罕见病领域进步与发展的必要条件。
3 中国罕见病研究的未来
未来,中国罕见病研究需秉承协作、创新的原则,从以下几个方面入手,推进罕见病事业发展。
3.1 构建协同创新网络
罕见病不仅是医疗难题,也是社会难题,需要政策制定者、医疗机构、产业界及个人等利益相关方共同形成协作配合体系。罕见病研究难度大、投入高,政府主导的法律法规制订、科研项目资助及社会保障体系建设项目,将有效鼓励罕见病研究和诊疗活动的开展。在医疗机构间搭建全国性或区域性的罕见病注册登记平台,打通信息交流障碍,借助各方研究专长,有利于克服研究中的技术和操作难题,共同推进罕见病研究。
3.2 建设国家级罕见病数据库与知识库
生物组学与临床表型组学的关联研究一直以来是罕见病研究的重点,组学库的建立为发现重要发病机制提供了关键保障。利用高通量方法进行生物组学数据采集、标准化术语体系获取表型组信息,并结合大数据、高性能计算等技术分析数据,是数据库建设及应用的重点。知识库的建设将为罕见病的临床诊疗提供权威有效的参考,对提高疾病的诊治能力具有重大意义。中国罕见病临床队列研究将搭建多组学临床数据库与多中心生物样本库,还将拓展与包括Orphanet、中文版Genereviews、GeneCards和MalaCards等国内外重要知识库的合作关系,搭建适用于中国的罕见病知识库。
3.3 推进技术创新探索
技术进步是罕见病研究与诊疗的重要推动力量,高通量测序技术、多组学信息采集技术、大数据分析处理技术为罕见病大规模研究及准确诊断提供保障,而罕见病药物研发和以基因治疗为代表的生物治疗技术,是罕见病有效干预的关键。目前FDA批准了首个基于CAR-T技术的基因疗法,用于治疗急性淋巴细胞白血病[20],而在非肿瘤领域,RPE65相关性遗传性视网膜萎缩的基因疗法已获得FDA优先评审资格[21]。罕见病药物的适应证可能拓展至常见疾病的治疗,惠及多种常见疾病患者。罕见病队列研究将为各类技术提供研究平台,促进技术成熟转化,最终更好地实现罕见病的诊断治疗,造福患者。
3.4 促进专业人才培养
罕见病的研究与诊疗中,人才的充分参与起到关键性作用,罕见病的充分研究与干预需要多方位多层次人才参与。在罕见病人才队伍中,将包括临床医生及遗传咨询人才、科研及数据分析人才、政策制定及社会科学人才等多类型人才。引入合作机制,鼓励人才协作创新,将全方位推动罕见病事业的进步。而开展高水平、大规模的罕见病研究,既是人才施展才华的最佳平台,也是培养人才的可靠途径。
3.5 推动患者关怀
现代医学的治疗方案与药物不能完全治愈疾病,但患者关怀可以涵盖整个病程。良好的医患关系、深入的患者教育、积极的心理干预及有力的社会保障是患者关怀的重要组成部分。通过以上几方面的努力,能够让患者积极参与疾病管理并充分融入日常社会生活。同时,在罕见病队列构建的过程中,患者组织将推进联络机制、互助机制与教育机制的建设,鼓励和支持患者更主动地参与罕见病注册研究。
3.6 搭建国际合作平台
罕见病不仅仅是中国的挑战,也是世界的挑战,中国在罕见病领域作出的努力,也将造福全球患者。通过广泛的国际合作,中国罕见病研究将对接先进的国际经验与研究技术,参与国际研究项目,推进国际罕见病事业发展。中国国家罕见病注册体系将通过与国际协作网络的协同努力,促进国内外资源共享,共同应对罕见病全球挑战。
4 小结
罕见病诊治的研究是挑战,也是未来医学发展重大突破的方向。中国是人口大国,同时也是罕见病疾病负担大国,以NRDRS为代表,将集中罕见病病例资源,积极推动多方协作、鼓励创新发展,努力快速提高罕见病诊断水平、强化罕见病治疗手段、提高对患者的服务能力,为罕见病事业的发展与人类健康作出应有的贡献。
[1] Regulation (Ec) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on Orphan Medicinal Products [J]. OJEC, 2000, 43: 1- 5.
[2] Rare Diseases Act of 2002 [Z]. US Congress, 2002.
[3] Boycott KM, Vanstone MR, Bulman DE, et al. Rare-disease genetics in the era of next-generation sequencing: discovery to translation [J]. Nat Rev Genet, 2013, 14: 681- 691.
[4] Therapeutic Goods Regulations 1990 [Z]. 1990.
[5] Japan pharmaceutical manufacturers association information on Japanese regulatory affairs[R].2017.http://www.phar-mexcil.org/v1/docs/Japan_pharmaceutical_Administration.pdf.
[6] Orphanet. Rare disease registries in Europe[R]. 2017. https://www.orpha.net/orphacom/cahiers/docs/GB/Registries. pdf.
[7] Boycott KM, Rath A, Chong JX, et al. International cooperation to enable the diagnosis of all rare genetic diseases [J]. Am J Hum Genet, 2017, 100: 695- 705.
[8] U.S. Food & Drug Administration. 2016 Novel drugs summary [R]. 2017. https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM536693.pdf.
[9] Evaluate Pharma. Evaluate Pharma Orphan Drug Report 2017[R]. 2017. http://info.evaluategroup.com/rs/607-YGS- 364/images/EPOD17.pdf.
[10] Wu N, Ming X, Xiao J, et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis [J]. N Engl J Med, 2015, 372: 341- 350.
[11] Sun L, Zhao L, Du L, et al. Identification of two novel mutations in the GALNT3 gene in a Chinese family with hyperphosphatemic familial tumoral calcinosis [J]. Bone Res, 2016, 4: 16038.
[12] Wu W, Lu CX, Wang YN, et al. Novel phenotype-genotype correlations of restrictive cardiomyopathy with myosin-binding protein c (Mybpc3) gene mutations tested by next-generation sequencing [J]. J Am Heart Assoc, 2015, 4: e001879.
[13] Ding F, Wickman L, Wang SQ, et al. Accelerated podocyte detachment and progressive podocyte loss from glomeruli with age in Alport Syndrome [J]. Kidney Int, 2017.doi: 10.1016/j.kint.2017.0.017. [Epub ahead of print].
[14] Hou Y, Yang J, Luo C, et al. Patterns of striatal functional connectivity differ in early and late onset Parkinson’s disease [J]. J Neurol, 2016, 263: 1993- 2003.
[15] Wu WH, Yang L, Peng FH, et al. Lower socioeconomic status is associated with worse outcomes in pulmonary arterial hypertension [J]. Am J Respir Crit Care Med, 2013, 187: 303- 310.
[16] Xu KF, Lo BH. Lymphangioleiomyomatosis: differential diagnosis and optimal management [J]. Ther Clin Risk Manag, 2014, 10: 691- 700.
[17] 中共中央办公厅, 国务院办公厅.《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》[Z]. 2017.
[18] 冯时, 弓孟春, 张抒扬. 中国国家罕见病注册系统及其队列研究:愿景与实施路线 [J]. 中华内分泌代谢杂志, 2016, 32: 977- 982.
[19] 肖磊. 患者组织在罕见病和孤儿药研发中的作用 [J]. 国际药学研究杂志, 2017, 2: 209- 214.
[20] FDA approval brings first gene therapy to the United States [N]. 2017.https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm574058.htm.
[21] Humphrey M. FDA Consider Approval of US Gene Therapy [N]. 2017.http://www.frontlinegenomics.com/news/13504/fda-consider-approval-of-us-gene-therapy/.
猜你喜欢
疯狂英语·新读写(2021年10期)2021-12-07 02:41:30
中华养生保健(2020年5期)2020-11-16 01:43:52
奥秘(2019年8期)2019-08-28 01:47:05
现代园艺(2017年21期)2018-01-03 06:41:32
商周刊(2017年7期)2017-08-22 03:36:21
小猕猴智力画刊(2016年6期)2016-05-14 09:21:40
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国医药科学(2015年15期)2015-02-27 12:32:25
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
