
肌萎缩侧索硬化的临床分型、分期及病情评估
2018-02-02 09:15:33李晓光刘明生崔丽英
协和医学杂志 2018年1期
李晓光,刘明生,崔丽英
中国医学科学院 北京协和医学院 北京协和医院神经科, 北京 100730
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)是一种进展性神经系统变性疾病。由于上、下运动神经元丢失导致球部、四肢、胸部肌肉逐渐无力和萎缩,动眼肌及括约肌不受累。其起病隐袭,进展缓慢,最终因呼吸衰竭死亡。发病率约1.5/10万,患病率4~6/10万。发病年龄平均55岁,自发病起平均存活3.5年,50%患者平均存活期为2.5年,5年存活率20%,10年存活率10%。球部起病者存活时间约2.2年,很少超过5年。一般发病年龄越早,存活时间越长[1]。目前尚无治愈方法,近年来研究表明多种措施可延长患者存活期,提高患者生活质量。为了科学合理地治疗ALS,本文主要介绍其临床分型、分期、治疗模式及病情进展评估和随访方法。
1 肌萎缩侧索硬化的命名
ALS在不同国家有不同名称,在法国称“Charcot病”,是为纪念1869年首次描述这一疾病的马丁·夏科医生而命名;在英国称“运动神经元病”,强调该病归属的类别;在美国称“Lou Gehrig病”,著名棒球手Lou Gehrig曾患此病并引起公众关注,因此而命名;专业杂志遵循国际神经病学联盟的命名称为“ALS”。我国一般将“ALS”和“运动神经元病”混用,近年来国内非医学媒体还称其为“渐冻人症”,源于台湾运动神经元病协会进行科普宣教所使用的俗称[2]。笔者建议国内同行统一采用ALS为该病的病名,便于和国际交流。
2 诊断
1990年,世界神经病学联合会在西班牙埃斯科里亚尔(El Escorial)举办的一次研讨会,制定了第一个共识文件[3],其核心是将病变累及的神经系统区域分为颈区、胸区、腰骶区、延髓区。根据制定的标准,在确诊ALS过程中需要同时存在上、下运动神经元变性临床表现并逐步向身体其他区域扩展、且无其他疾病可以解释其症状。电生理检查可用来确认某一区域是否存在下运动神经元损害,如每个区域至少有两个不同脊神经根和周围神经支配的肌肉同时存在纤颤电位,长时程巨大运动单位电位及运动单位募集减少。根据神经功能缺损的广泛性,EL Escorial标准制定了4个诊断信度级别:(1)肯定的ALS(在3个区域同时存在上、下运动神经元损害表现);(2)很可能的ALS(两个区域存在上、下运动神经元损害表现,有些上运动神经元表现位于下运动神经元表现的头端);(3)可能的ALS(一个区域存在上、下运动神经元损害的表现或至少两区域存在上运动神经元损害表现);(4)可疑的ALS(至少两个区域存在下运动神经元损害表现)。
1998年,修订标准(或Airlie House标准)[4]认为,在腰骶和颈椎区域不同脊神经根和周围神经支配的至少两块肌肉和在胸椎和头颅区域至少一块肌肉存在急、慢性失神经支配的表现,电生理检查可确定下运动神经元损害。患者两个区域存在下运动神经元损害表现,在一个区域存在上、下运动神经元表现或一个区域仅有上运动神经元表现时,即为临床实验室支持的“很可能的ALS”级别,故修订的El Escorial标准保留3个诊断信度级别:临床肯定的ALS、很可能的ALS、可能的ALS。去除了“可疑的ALS”级别。
因灵敏度低,且从发病到符合参加临床试验的资格需要较长时间,两套诊断标准一直备受争议。2006年,在日本淡路市召开的共识会议进行了两项修订:(1)在确认失神经时,下运动神经元损害的电生理证据(活动性及慢性去神经征)等同于临床的下运动神经元表现,束颤电位等同于纤颤电位和正锐波。从本质上讲,针电极肌电图被认为是一种临床检查的延续,并维持了前一版诊断标准的一般原则。其结果使临床实验室支持的“很可能的ALS”这一级别变得多余,故进一步简化了诊断标准。(2)要特别重视针肌电图对慢性神经性变化和肌束震颤的评估,留意无纤颤或正锐波的肌肉,其可改善ALS早期诊断的灵敏度,这对及早诊断、提高进入临床试验的时机和人数、减少延迟诊断、避免延误治疗等具有重要意义[5]。
目前ALS的诊断信度级别沿用1998年修改的EL Escorial标准,即分3级:临床肯定的ALS、很可能的ALS、可能的ALS。
3 临床分型
ALS通常分为经典型ALS、进行性延髓(球)麻痹、脊髓性肌萎缩和原发性侧索硬化4种临床亚型。但这4种亚型难以准确概括所有的病情发展与损害分布特点,因此建议采用新的ALS分型[6],以利于判断患者预后及在临床药物试验中选择合适患者入组。新的分型主要将ALS分为8种临床表型,在发病年龄、延迟诊断时间、合并额颞叶痴呆(fronto temporal dementia,FTD)比率、生存期及3、5、10年存活率等方面均有差异(表1)。
3.1 经典型ALS
在上肢或下肢出现特征性症状或体征,锥体束征明确,但并不突出。
3.2 延髓型ALS
此类患者为延髓发病,有构音障碍和/或吞咽困难、舌萎缩、肌束震颤。在发病后6个月内无脊髓损害症状;在发病后前6个月锥体束征可不明显,但之后显而易见。
3.3 连枷臂综合征
特点主要是上肢近端无力和萎缩,逐渐发展。此类型包括病程中某一阶段患者上肢的病理性深部腱反射或霍夫曼征,但无肌张力增高或阵挛。在发生症状后局限于上肢的功能受累至少持续12个月。
3.4 连枷腿综合征
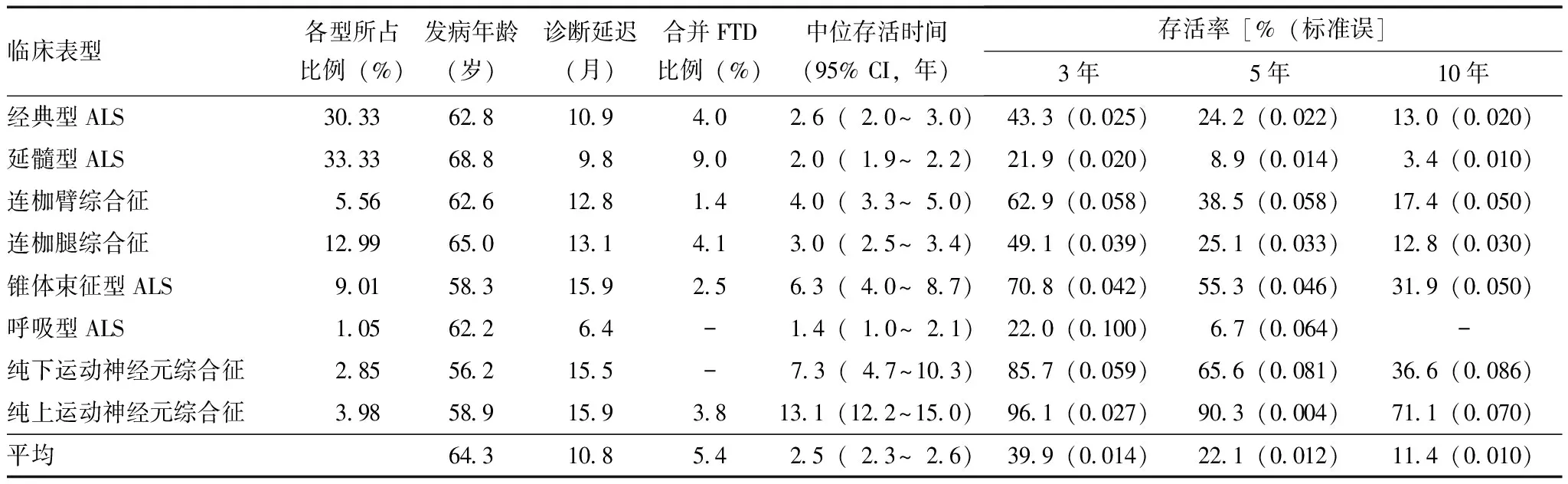
表 1 肌萎缩侧索硬化临床表型与存活预测[6]
FTD:额颞叶痴呆
特点是下肢远端无力和萎缩逐渐进展,包括病程中某一阶段患者下肢的病理性深部腱反射或巴彬斯基征,但无肌张力增高或阵挛。患者下肢近端起病的萎缩和无力,在无远端受累时列为经典型ALS。
3.5 锥体束征型ALS或上运动神经元损害突出的ALS
临床表现主要为锥体束征,严重的痉挛性截瘫/四肢瘫,有一个或多个以下体征:巴彬斯基征、霍夫曼征、腱反射亢进、下颌阵挛性抽动、构音障碍和假性球麻痹。痉挛性麻痹可存在于发病初期或疾病晚期,发病时至少在两个不同区域同时表现出明显的下运动神经元损害体征,如肌肉无力和萎缩,肌电图检查存在慢性和活动性的失神经损害。
3.6 呼吸型ALS
发病时表现为弥漫性呼吸功能损害,休息或劳累时端坐呼吸或呼吸困难,在发病第6个月后只仅轻微脊髓或延髓体征,可有上运动神经元受累的表现。
3.7 纯下运动神经元综合征
有逐渐进展的下运动神经元受累的临床表现和电生理证据。此类型需排除以下6种情况:(1)以标准化神经节段传导研究存在运动传导阻滞者;(2)临床上有上运动神经元受累体征者;(3)类运动神经元病综合征疾病史者;(4)有家族病史的脊髓性肌萎缩症;(5)SMN1基因的缺失者,或由雄激素受体基因重复异常扩展导致的遗传性延髓脊髓性肌萎缩症;(6)神经影像学研究有结构损害者。
3.8 纯上运动神经元综合征
上运动神经元损害的临床症状包括严重的痉挛性截瘫/四肢瘫、巴彬斯基征或霍夫曼征、反射极度活跃、下颌阵挛性抽动、构音障碍和假性球麻痹。类型需排除以下4种情况:(1)随访过程中按照西班牙EL Escoriad标准有临床或肌电图表现的下运动神经元受累表现者;(2)类运动神经元病综合征病史者;(3)有痉挛性截瘫/四肢瘫家族史者;(4)有基因突变相关遗传性痉挛性截瘫(SPG3A、SPG4、SPG6、SPG7和SPG20)者。
以上8种表型建立在诊断时临床表现的基础之上,但在随访中要收集患者所有可用的资料,不断修订。
4 临床分期
为了相对客观准确判断ALS的预后,对疾病发展情况进行划分以利于判断药物临床试验的效果及对不同阶段患者进行有益干预。至今已报道的分期方法有3种。
英国Roche等[7]根据病程中患者最近一次受累区域的不同,将ALS分为5期(即伦敦分期):1期为出现症状(第一区域受累);2期A为确诊,2期B为第二区域受累;3期为第三区域受累;4期A为患者需要经皮胃肠造瘘(percutaneous endoscopic gastrostomy, PEG),4期B为患者需要无创通气;5期为死亡或进行机械通气。各期平均存活时间及5年生存率均不同(表2)。
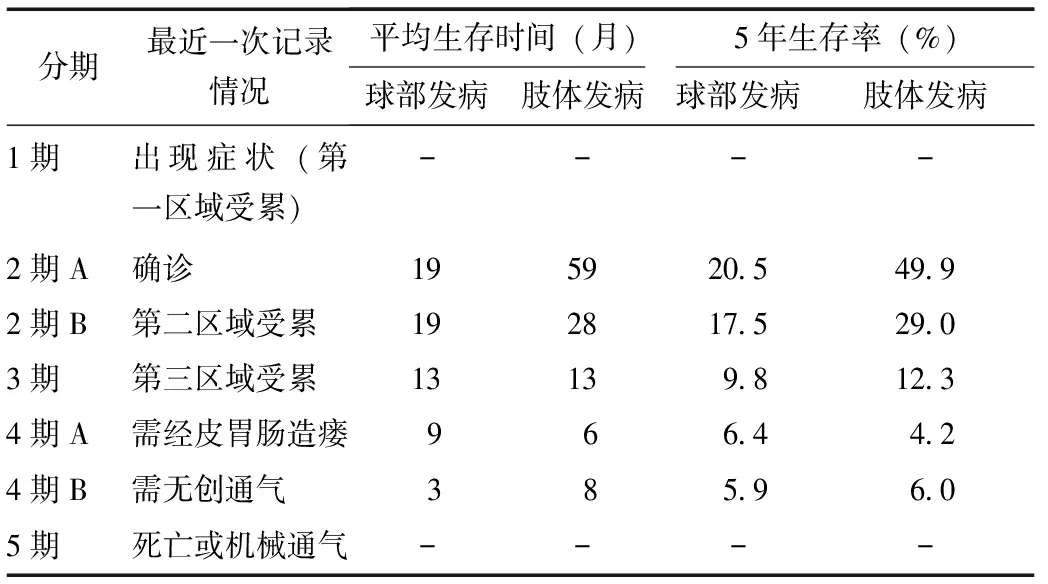
表 2 肌萎缩侧索硬化伦敦分期与生存期
意大利Chiö等[8]提出的米兰都灵ALS分期系统(Milano Torino functional staging,ALS-MITOS),将患者功能分为4个域,通过将4个关键点(包括活动、吞咽、交流和呼吸)、换算为0(低于阈值)或1(高于阈值),根据每个域功能评分总和来确定分期:0期为无功能丧失;1期为一个功能域丧失;2期为两个功能域丧失;3期为3个功能域丧失;4期为4个功能域丧失;5期为死亡或进行机械通气(表3)。Chiö等通过前瞻性研究,按ALS-MITOS分期系统观察了118例ALS患者12个月的病程进展,发现分期与生活质量、神经功能、医疗费用密切相关,且还可预测患者生存时间[9]。
这两个分期系统各有优点。伦敦分期基于临床病程节点(如起病症状、诊断及后续进展等),验证了地区ALS专科中心的1471例患者;ALS-MITOS分期系统基于功能评定量表(ALS functional rating scale, ALSFRS),是大多数ALS专家熟悉的评分量表,结合与患者功能相关的临床节点(如诊断,第二、三区域受累)或两个特定功能的丧失(如需PEG或无创通气),广泛应用于药物临床试验和日常工作。
在这两个分期系统之前,曾有一个以ALS康复治疗为目的的分期系统,其不能反映患者的病程进展情况,也无法应用于疾病早期[10],故不作详细介绍。
5 治疗模式
ALS治疗分5种类型,即预防、治愈、阻止疾病进展、减缓疾病进展及减轻疾病影响。预防ALS发生,要确定病因,避免疾病发作,目前遗传性ALS可做到,前提是找到明确的致病基因。治愈(使疾病的损害逆转)或阻止疾病发展(不能使疾病的损害逆转)目前尚不能实现,目前临床上仅能做到有限减缓病情发展或延长生命,减轻疾病带来的痛苦和不便。谷氨酸释放抑制剂已在ALS患者及转基因动物治疗中取得较好疗效,是目前唯一通过美国食品药品监督管理局认证可有效延缓病情进展并在全球广泛使用的药物[11]。无创正压通气(non-invasive positive pressure ventilation,NIPPV)和PEG也可延长患者的存活,提高患者生活质量[12]。在减轻疾病影响方面,需要取得社会及家庭支持,包括对受损特定功能的康复训练、抗抑郁治疗、并发症的治疗、构音障碍、流涎、咽下困难痉挛、疼痛及便秘的处理等,对提高患者生活质量具有重要意义[13]。

表 3 肌萎缩侧索硬化米兰都灵分期评分[8]
域功能评分标准:将肌萎缩侧索硬化进展的关键点(活动、吞咽、交流和呼吸4个功能域)评分换算为0(低于阈值)或1(高于阈值),将4个域功能评分的数值相加确定分期,如两项均用,得分基于两个项目的总分 NIPPV:无创正压通气
6 临床评估
ALS目前尚无治愈办法,准确、客观、科学评估病情及神经功能状态对了解疾病进展程度和药物疗效极为重要。
6.1 存活
将ALS患者的存活作为药物临床试验观察指标无疑是合理的[14],但随着有创或无创通气支持技术及营养支持的发展使这一指标受到质疑。越来越多的证据显示NIPPV和PEG可以延长患者生命。因此,如将存活作为终点观察指标,设计新药的临床观察方案必须考虑NIPPV和PEG的因素。
6.2 运动系统特定损害的测定
手工运动测定肌力评分虽然在临床应用广泛,但受主观因素影响且无法标准化,因此推荐用定量肌力仪进行肢体肌力测定。球部功能尚无理想的评估办法,目前主要采用综合量表。前角细胞功能可用电生理对运动单位数目进行评估。上运动神经元功能常依据电生理测定(如经颅磁刺激)和功能性影像学测定(如磁共振波谱)结果进行评估[15]。
6.3 综合测定
综合测定通常应用综合量表进行评估,目前常用的有ALSFRS及生活质量量表(amyotrophic lateral sclerosis assessment questionnaire- 40,ALSAQ- 40)等。
6.3.1 ALSFRS
在ALS严重程度量表和帕金森统一评估量表基础上设计。增加了更多运动功能项目,主要由4个球部呼吸功能、两个上肢功能(使用餐具和穿衣)、两个下肢功能(走路和爬楼梯)及两个其他功能(穿衣及洗漱、床上翻身)组成,评分从0(不能完成任务)至40分(正常)。其优点是简便、容易操作、应用广泛,其敏感性、可靠性和稳定性已得到广泛确认,和已有的其他评估量表可比性和相关性强。其缺点是对呼吸功能评价比重较小,但设计者已认识到这一点,对量表进行了改良,增加了呼吸功能的评分强度,还适用于呼吸支持的使用者。目前改良的ALS功能评分量表总分48分[16]。
目前基于书面的ALSFRS版本应用广泛,且已开发了网络及电话版本,均得到验证,患者自评得分与医生评分高度一致,这是因为书面ALSFRS版本医生评分均基于患者对自身神经功能的描述[17]。Prize4组织基于ALSFRS斜度变化预测模型的外包竞赛,结果13组参赛者中两组胜出,优于组织者开发的预测模型,与参加临床试验的患者病情实际进展高度一致。组织者认为此模型可以降低20%的临床药物试验入组患者人数,节省大量费用[18]。
6.3.2 ALSAQ- 40
ALS自我评估问卷是在缺乏ALS特异生活质量量表的情况下,由牛津大学于1999年设计完成[19]。其指标包括身体运动能力、饮食能力、社会交往能力和情绪反应,优点是简单、可操作性强,可用于患者自我评价生活质量,便于信函随访。
6.4 呼吸功能测定
建议采用用力肺活量(forced vital capacity, FVC)进行评估,FVC是最重要的生存率预测指标。Fallat发现FVC<50%预计值提示预后不良,Bensimon等在利鲁唑临床试验中证实FVC可作为生存率预测指标,多年来FVC<50%在临床及试验研究中广泛用作决策的主要指标,美国神经病学会及欧洲神经病学会联盟的ALS治疗指南均将其列为NIPPV的使用指征[12- 14]。
6.5 生化指标
目前ALS尚无公认的诊断特异性生物学标志物和确定疾病进展的特异指标[20]。肌酸激酶、肌酐、同型半胱氨酸、超敏C反应蛋白、抑胱素C和尿酸可能和疾病进展有关,值得临床进一步研究。
7 小结
ALS目前尚无治愈办法,大量研究表明利鲁唑、PEG、NIPPV及对症支持对延长患者存活期,提高生活质量有积极作用。明确诊断后,进一步细分临床表型,确定临床分期,客观评估患者综合情况,有利于合理选择治疗方案,为新药临床试验提供客观的评估指标。
[1] Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis[J]. N Engl J Med, 2017, 377:162- 172.
[2] 李晓光,崔丽英.肌萎缩侧索硬化手册[M]. 北京:中国协和医科大学出版社,2013: 294.
[3] Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis[J]. J Neurol Sci, 1994,124:96- 107.
[4] Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis[J]. Amyotroph Lateral Scler Other Motor Neuron Disord, 2000,5:293- 299.
[5] de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS[J]. Clin Neurophysiol, 2008, 119:497- 503.
[6] Chiò A, Calvo A, Moglia C, et al. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study[J]. J Neurol Neurosurg Psychiatry, 2011, 82:740- 746.
[7] Roche JC, Rojas-Garcia R, Scott KM, et al. A proposed staging system for amyotrophic lateral sclerosis[J]. Brain, 2012, 135:847- 852.
[8] Chiò A, Hammond ER, Mora G, et al. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis[J]. J Neurol Neurosurg Psychiatry, 2015, 86:38- 44.
[9] Tramacere I, Dalla Bella E, Chiò A, et al. The MITOS system predicts long-term survival in amyotrophic lateral sclerosis[J]. J Neurol Neurosurg Psychiatry, 2015, 86:1180- 1185.
[10] Sinaki M, Mulder DW. Rehabilitation techniques for patients with amyotrophic lateral sclerosis[J]. Mayo Clin Proc, 1978, 53:173- 178.
[11] Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis(ALS)/motor neuron disease(MND) [J]. Cochrane Database Syst Rev, 2012, 3:CD001447.
[12] Andersen PM, Abrahamsb S, Borasio D, et al. EFNS guidelines on the Clinical Management of Amyotrophic Lateral Sclerosis(MALS)—revised report of an EFNS task force[J]. Eur J Neurol, 2012, 19:360- 375.
[13] Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies(an evidence-based review):report of the Quality Standards Subcommittee of the American Academy of Neurology [J]. Neurology, 2009, 73:1218- 1226.
[14] Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS /Riluzole Study Group[J].N Engl J Med, 1994, 330:585- 591.
[15] Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis[J]. Nat Rev Neurol, 2011, 7:639- 649.
[16] Cedarbaum JM, Stambler N, Malta E,et al. The ALSFR-SR: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group(Phase III) [J]. J Neurol Sci, 1999,169:13- 21.
[17] Maier A, Holm T, Wicks P, et al.Online assessment of ALS functional rating scale compares well to in-clinic evaluation: a prospective trial[J]. Amyotroph Lateral Scler, 2012,13:210- 216.
[18] Küffner R, Zach N, Norel R, et al. Crowdsourced analysis of clinical trial data to predict amyotrophic lateral sclerosis progression[J]. Nat Biotechnol, 2015,33:51- 57.
[19] Jenkinson C, Levvy G, Fitzpatrick R, et al. The amyotrophic lateral sclerosis assessment questionnaire(ALSAQ- 40): tests of data quality, score reliability and response rate in a survey of patients[J]. J Neurol Sci, 2000, 180:94- 100.
[20] Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis[J]. Nat Rev Neurol, 2014,10:661- 670.
猜你喜欢
考试与评价·高二版(2020年2期)2020-09-10 07:22:44
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:48
中外医疗(2015年16期)2016-01-04 06:51:42
实用手外科杂志(2015年1期)2015-08-27 01:52:06
质量与标准化(2015年9期)2015-07-10 15:12:07
西安交通大学学报(医学版)(2015年2期)2015-02-28 17:59:20
浙江人大(2014年5期)2014-03-20 16:20:25
中医研究(2013年9期)2013-03-11 20:27:37
世界科学(2013年11期)2013-03-11 18:09:46
中国合理用药探索(2011年9期)2011-03-20 16:30:23
