
黑色素瘤的生物标志物:从基因组学到表观遗传学
2018-02-02 09:15吴海竞付思祺李倩文张慧明陆前进
协和医学杂志 2018年1期
吴海竞,付思祺,李倩文,张慧明,陆前进,郭 重,2,3
1中南大学湘雅二医院皮肤科 湖南省表观遗传重点实验室,长沙 410011 哈佛大学布莱根妇女医院 2皮肤病理科 3病理科,波士顿,美国 02115
黑色素瘤是一种恶性程度高且生存率低的皮肤肿瘤,发病速度快且容易转移至脑、肝脏和肺等人体重要器官。目前对于黑色素瘤的临床诊断主要基于组织病理学标准,包括肿瘤深度、侵袭水平、是否有溃疡和淋巴结转移。但组织病理学标准无法区分从良性黑色素痣转变为黑色素瘤的亚型,亦无法预判哪些患者容易发生转移,因此,目前研究重点为尽快找到一种可用于黑色素瘤早期诊断和预测转移可能性的新型且有效的生物标志物。此外,虽然目前化学疗法和免疫疗法发展迅速,如威罗菲尼/拉菲尼 (BRAF 抑制剂)[1- 4], 纳武单抗(PD- 1抗体)[5],易普利姆玛(CTLA- 4 抗体)[6]以及派姆单抗[7]等药物,可延长患者生存期,但大多数患者最终发生耐药,预后不佳。因此,可预测药物反应性的生物标志物可能实现针对黑色素瘤的个体化和靶向治疗,为黑色素瘤的有效控制带来曙光。
迄今为止,已发现一系列新的生物标志物,其中一些可预测黑色素瘤转移、药物反应性等,甚至有些可作为治疗靶点。本文将从基因组学到表观遗传学水平,对新发现的生物标志物进行全面系统总结,讨论生物标志物临床应用的可行性,为改善患者生活质量、延长患者生存期等提供潜在的实验室指标和治疗靶点。
1 黑色素瘤的基因生物标志物
二代测序技术(next-generation sequencing,NGS)的发展成功帮助科学家们发现疾病新的潜在致病基因。事实上,目前基因分析已投入临床应用。例如,在应用抑制BRAF药物治疗之前,医生会应用基因分析确定肿瘤细胞的BRAF 基因是否发生突变。一项来自TCGA的研究从331例患者333份黑色素瘤标本中提取了DNA、RNA和蛋白质,并绘制出癌症基因图谱,构建了不同突变基因(BRAF、NRAS、KIT、GNAQ/GNA11、TP53/CDKN2A和NF1)的分类体系[8],可指导治疗策略的制定(表1)。从全基因组测序(whole-genome sequencing,WGS)和全外显子测序(whole-exsome sequencing,WES)的研究中发现,黑色素瘤患者存在高突变率及高紫外线暴露率的特征,而紫外线照射正是黑色素瘤的高危风险因素[8- 10]。通过Bonfferoni统计学方法检验(P<0.05),癌症基因图谱研究新发现了13个黑色素瘤致病基因,分别为BRAF、 NRAS、TP53、NF1、CDKN2A、ARID2、 PTEN、PPP6C、RAC1、 IDH1、DDX3X、MAP2K1和RB1[8],然而,一些经典的导致其他部位癌症的致病基因如PREX2、GRIN2A、ERBB4、ADAMTS18、BCL2L12、SOX10、MITF和KIT, 却未在此癌症基因图谱中出现,可能由不同的统计学方法差异所导致[11]。
基因除了可作为生物标志物用于肿瘤诊断,基因突变还可预测其转移可能性及对靶向药物治疗抵抗的概率。例如,NMDAR2 和 EGFR4 突变、MET 和MITF表达增加以及PTEN表达丢失预示转移可能性大,而MITF 和BRAF表达增加及PTEN表达丢失则预示可能对靶向治疗抵抗。NRAS突变是目前发现的唯一一个可预测免疫治疗敏感性的基因标志物[23]。这些证据表明基因分析对监控疾病进展和指导个体化治疗具有重要作用。
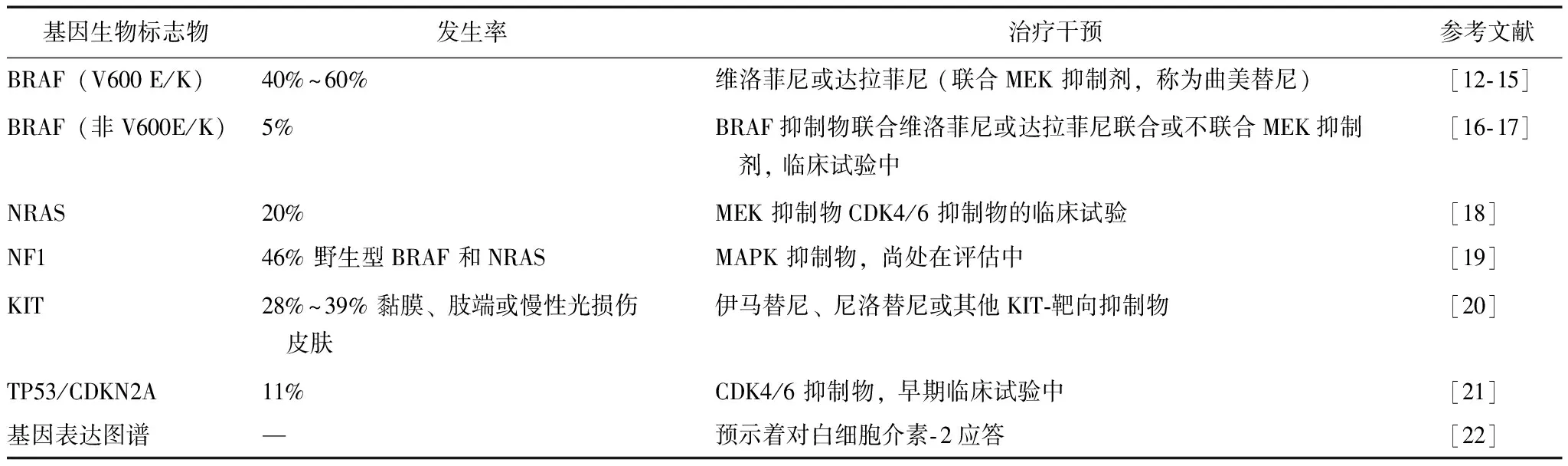
表 1 黑色素瘤靶向治疗的基因生物标志物
除了这些研究较多的基因外,其他候选基因也有可能作为生物标志物。如在既往研究中,有学者试图将整合素信号通路基因的单核苷酸多态性(single nucleotide polymorphisms,SNPs)与皮肤黑色素瘤存活率相关联。德克萨斯大学M.D.安德森癌症中心的全基因组相关性研究发现,整合素基因通路中3个独立的SNPs:DOCK1 rs11018104 T>A、 rs35748949 C>T和PAK2 rs1718404 C>T有望成为预测黑色素瘤预后的生物标志物[24];在肢端雀斑样黑色素瘤中发现TERT变异,TERT抑制物可能成为潜在治疗策略,使得TERT变异成为制定黑色素瘤个体化治疗策略的潜在生物标志物[25];从33例葡萄膜黑色素瘤患者中的WGS研究发现,葡萄膜黑色素瘤的可能致病基因为GNAQ、GNA11、BAP1、 EIF1AX和 SF3B1, 并发现了体细胞突变中的其他基因如TP53BP1、CSMD1、TTC28DLK2及KTN1[26],但仍需进一步扩大样本量确定这些基因突变是否与葡萄膜黑色素瘤的发生发展具有紧密联系。
2 黑色素瘤的表观遗传学生物标志物
表观遗传学是指在基因表达和功能中不涉及DNA核苷酸原始序列改变的潜在遗传学改变,可最终决定基因表达或沉默,因此在细胞分化、生长、发育、老化和免疫反应等生命过程中发挥了关键作用。表观遗传学为环境因素如何决定个体表型差异提供了除遗传学以外的解释,同时也为一些疾病如黑色素瘤除遗传易感性以外的发病机制提供了证据。随着表观遗传学时代的到来,已发现越来越多表观遗传学生物标志物涉及DNA甲基化/羟甲基化、组蛋白修饰和非编码RNA[微小RNA (micro-RNA, miRNA)和长链非编码RNA(long non-coding RNA,lncRNA)]。对比靶向治疗的基因生物标志物,表观遗传学治疗具有可控、可逆等优点。
2.1 DNA甲基化/羟甲基化
在许多真核物种中,DNA甲基化是相对稳定并可遗传的表观遗传学标志物,其是指甲基添加到腺嘌呤或胞嘧啶中嘧啶环的第5个位置,使得胞嘧啶成为甲基胞嘧啶。DNA高甲基化代表基因表达受到抑制,因此涉及很多生命过程,如细胞分化与增殖。DNA甲基化由甲基化转移酶如DNMT1、 DNMT3a 和DNMT3b等介导。每一种甲基化转移酶的功能有所差异,如DNMT1在细胞复制过程中维持甲基化状态,而DNMT3a和DNMT3b常常诱导甲基化的初始化过程。相反,DNA去甲基化是甲基化修饰丢失的过程。DNA甲基化消除主要涉及两种途径:一是DNA被动去甲基化, 即在 DNA 复制时,DNA 甲基化模式的维持受到干扰,未保留原有甲基化模式,随着复制的进行,甲基化的CpG被“稀释”,导致 DNA 去甲基化;二是DNA主动去甲基化,这一过程不依赖于DNA复制,而受酶的催化, 使5-甲基化胞嘧啶(5-methylated cytosine, 5mC)转化为未甲基化的胞嘧啶。DNA羟甲基化介导的主动去甲基化途径主要包括两种:(1)氧化去甲基化途径: 5-羟甲基化胞嘧啶(5-hydroxymethylated cytosine, 5-hmC)先被 TET 蛋白催化转变为 5-氟胞嘧啶(5-formylcytosine, 5fC),后者继续在TET蛋白的催化作用下转变为 5-羧基胞嘧啶(5-carboxylcytosine, 5caC),然后被胸腺嘧啶DNA糖基化酶(thymine DNA glycosylase,TDG)脱羧还原为胞嘧啶[27];(2)另外一条途径为5fC直接被TDG脱羧还原为胞嘧啶。这个过程中,5mC氧化成5-hmC是反应的第一步也是关键一步,这一步由TET家族(TET1、TET2、TET3)加双氧酶调节[28]。5-hmC是活性DNA去甲基化最丰富的中间媒介物,在正常发育和癌症发生中发挥正转录调控作用[29],其含量直接与人体各个组织器官的分化水平相关[30]。
在早期研究中,肿瘤发生发展过程中发现全基因组DNA低甲基化[31]。有研究认为DNA低甲基化可促进早期肿瘤细胞增殖和后期转移,使其具有生存优势[32],且在着丝粒序列和重复序列方面与染色体不稳定相关[33]。另一方面,在启动子区CpG岛的DNA甲基化被认为通过沉默抑癌基因促进肿瘤发生。许多高甲基化的抑癌基因,参与生物学过程,包括细胞周期调控、DNA修复、细胞信号传导、基因转录和细胞凋亡,如已报道的其在黑色素瘤中作为生物标志物的潜在可能性[ 34]。
除CDKN2A, RAR-b2, RASSF1A和IDH1[35]等已被其他学者广泛研究的基因外,黑色素瘤中还发现其他高甲基化的基因。如在几项癌症研究中发现LINE- 1是全基因组甲基化的临床试验替代标志物,并在巴西的黑色素瘤患者中发现LINE- 1高甲基化,可能成为皮肤黑色素瘤的生物标志物[36]。在不同肿瘤类型中还发现LINE- 1甲基化状态与癌症风险因素相关[37]。此外, Claudin11被认为是潜在的诊断黑色素瘤的表观遗传学生物标志物[38],Claudin基因家族包含27个成员,其编码细胞间紧密连接的膜蛋白。肿瘤转移灶的位置与甲基化频率显著相关,表明原发性黑色素瘤的甲基化水平可能有助于判断黑色素瘤转移能力的差异。因此,研究Claudin11失活分析功能改变很有意义。此外,MGMT编码一种修复蛋白,鸟嘌呤残基的O6位置去除烷基,其启动子区甲基化状态已被提议作为胶质瘤[ 39 ]、大肠癌[ 40 ]和黑色素瘤[ 41 ]的生物标志物。在黑色素瘤患者的肿瘤和血清中发现MGMT基因的表观遗传沉默[42],提示其在肿瘤发生发展中的重要作用。MITF是另一个DNA高甲基化基因,一种控制细胞周期和黑色素生成的转录因子[43]。在不止一例肿瘤皮损和黑色素瘤患者外周血中发现MITF启动子区高甲基化。有趣的是,不同黑色素瘤标本和肿瘤[44]中MITF表达存在变化,其高表达代表高度增殖和分化,而相对低表达可能表明侵袭能力增强[45],提示MITF基因启动子高甲基化水平可能与疾病严重程度有关(表2)。当然,仍需更多研究进一步明确高甲基化基因在黑色素瘤中的作用,深入探索利用这些甲基化生物标志物和治疗靶点来治疗黑色素瘤。
事实上,运用定量甲基化特异性PCR检测黑色素瘤患者外周血中肿瘤相关基因的甲基化水平已在临床应用。例如,与对生物化学疗法不敏感的肿瘤相比,循环中较少甲基化的RASSF1A与生物化学疗法的敏感性相关,RASSF1A的甲基化水平与总体生存率相关[68];黑色素瘤基因组整体表现为低甲基化,更重要的是,5-hmC缺失可作为生物标志物来区分黑色素瘤与生理性黑色素细胞及良性增殖[69],且5-hmC缺失与黑色素瘤预后差具有很强相关性,预示5-hmC水平可作为潜在的预测预后的生物标志物,随后几年中的其他研究也进一步证实了该发现[70- 74];虽然在黑色素瘤中发现IDH2和TET蛋白水平降低,然而这种改变的上游和下游机制尚不清楚,TET蛋白已用于癌症临床前研究[75],如沉默TET2和TET3在上皮细胞向间质细胞转变过程和黑色素瘤转移中发挥重要作用[76]。
2.2 组蛋白修饰
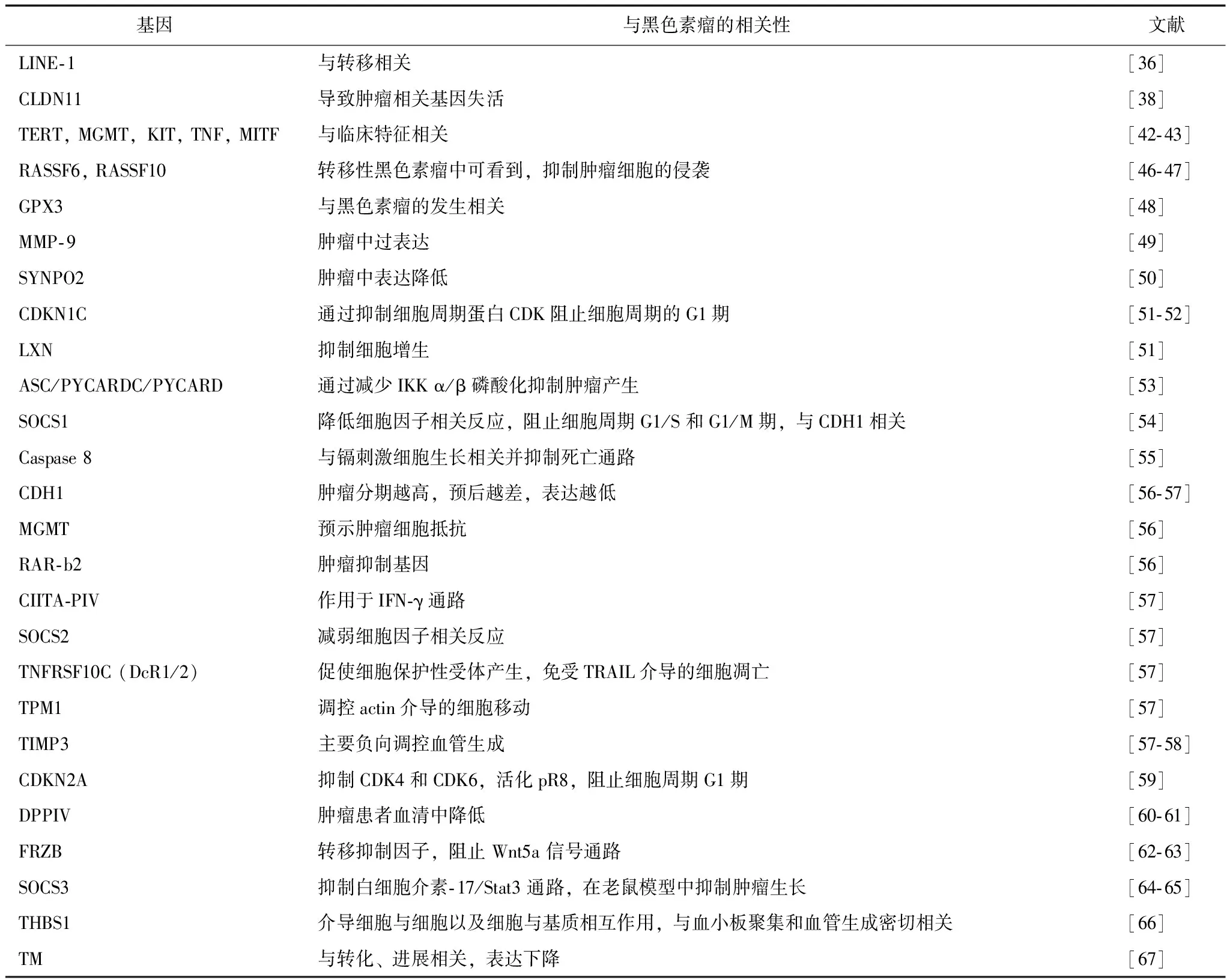
表 2 黑色素瘤中高甲基化的基因
组蛋白修饰是调控基因表达的另一重要表观遗传机制。DNA被包装成细胞核作为染色质,核小体是染色质的基本亚单位。每个核小体由146个碱基对的DNA形成两圈缠绕在组蛋白核心及两对H2A、H2B、H3和H4上。组蛋白修饰表现为从核小体凸出的小蛋白尾巴被修饰,包括甲基化、乙酰化和泛素化。每个修饰均有其独特的功能,例如组蛋白H3K9乙酰化增强转录,而甲基化则抑制该过程。在这些修饰中,乙酰化和脱乙酰作用得到了深入研究,分别由组蛋白乙酰化转移酶(histone acetyltransferase, HAT)和组蛋白脱乙酰化酶(histone deacetylase,HDAC)催化。HAT将乙酰基转移至赖氨酸,导致基因活化;HDAC去除乙酰基,导致基因沉默。与乙酰化不同,组蛋白甲基化发生在精氨酸和赖氨酸残基并受组蛋白甲基转移酶(histone methyltransferase,HMT)和其他酶的调节,且甲基化的影响受修饰后残基位置和甲基基团数目的调节,如H3K4me3增加基因表达,而H3K9me3和H3K27me3则导致基因表达下降。
组蛋白的改变与黑色素瘤免疫逃逸具有一定关系。研究发现组蛋白修饰蛋白和组蛋白修饰酶的异常表达与黑色素瘤的异常增殖相关[77]。例如,组蛋白低乙酰化已被证明可减少黑色素瘤PI3K/Akt信号通路肿瘤抑制基因的表达[78],异常的组蛋白乙酰化调控肿瘤细胞和肿瘤浸润免疫细胞中凋亡途径TRAIL/Apo2L[79]和Bcl- 2家族成员(Bim、Bax和Bak)[80]的表达,均提示组蛋白修饰可作为生物标志物预测免疫疗法的反应或作为HDAC抑制剂的治疗靶点。
除了总体组蛋白修饰状态外,组蛋白修饰酶也与黑色素瘤的发展相关,并有潜力成为生物标志物和治疗靶点。例如,EZH2是多梳蛋白抑制复合体上的H3K27甲基转移酶催化亚单位。EZH2的表达与黑色素瘤中高增殖率和高侵袭性的肿瘤亚型相关,其通过催化H3K27的转录从而抑制甲基化[81]。此外, 48%的EZH2基因高表达患者具有5年总生存期,而71%的EZH2基因低表达患者具有5年总生存期[81],说明EZH2基因可预测黑色素瘤患者的生存期。此外,通过CDKN1A基因乙酰化和去乙酰化,EZH2耗尽与黑色素瘤细胞促凋亡作用相关[82]。基于黑色素瘤高发和易转移的特性,EZH2抑制剂可作为其治疗的评估方法[83]。最近一项研究中一种新的定量质谱分析确定了伴随EZH2高表达,出现组蛋白翻译后修饰H3K27me3。人类晚期黑色素瘤组织中已发现EZH2介导的RUNX3基因和肿瘤抑制基因E-cadherin的沉默[84]。另一个例子是SETDB1,在黑色素瘤斑马鱼模型中发现甲基化组蛋白H3赖氨酸9,可加速黑色素瘤的形成[85],半数以上的黑色素瘤患者样本中检测出SETDB1的过表达[86]及其表达与抑制肿瘤抑制基因p16 的表达相关[87]。
2.3 微小RNA
miRNA是21~25个碱基对的非编码RNA,其通过结合靶基因信使RNA3’未翻译区,使信使RNA裂解,抑制翻译或使翻译受阻。研究表明黑色素瘤不同类型细胞和组织中miRNA表达异常,因而可能辅助诊断、判断预后,甚至作为潜在的治疗靶点。与其他标志物不同,细胞死亡裂解后会释放miRNA进入循环,肿瘤细胞也可通过外泌体释放进入循环。
越来越多的证据表明,miRNA通过调节重要的通路或基因发挥癌基因或抑癌基因作用,这种通路或基因涉及细胞增殖、凋亡、迁移和侵袭,药物抵抗和血管生成,如miRNA- 29c和miRNA- 324- 3p在转移性黑色素瘤中不表达,但具有预测黑色素瘤转移的作用。事实上,已发现许多miRNA在黑色素瘤的病理生理过程中发挥重要作用,如miRNA- 15b、 miRNA- 99a、 miRNA- 137、 miRNA- 148、 miRNA- 149、miRNA- 193b、 miRNA- 211、 miRNA- 221 和miRNA- 506- 514参与细胞增殖和生长,miRNA- 18b、 miRNA- 26a、 miRNA- 34a、miRNA- 34b/c、miRNA- 137、 miRNA- 203 和 miRNA- 205参与细胞凋亡,miRNA- 214、miRNA30b/30d、miRNA- 182、 let- 7a、miRNA- 126、miRNA- 145、miRNA- 137、miRNA- 18b、 miRNA- 34a/c、 miRNA- 211、miRNA- 9和miRNA- 31参与细胞侵袭和转移,miRNA-1908、 miRNA- 199a- 5p 和 miRNA- 199a- 3参与血管生成,miRNA- 200c则参与药物抗性。
黑色素瘤中研究较多并具有诊断和治疗潜能的miRNA包括miRNA- 21、miRNA- 125b、miRNA- 155、miRNA- 205、miRNA- 211等。miRNA- 21在黑色素瘤中表达增加,通过靶向调节TIMP3、PDCD4、BCL- 2和PTEN,在黑色素瘤的发生发展中发挥重要作用[88- 89];miRNA- 21升高预示患者5年非进展生存期和总生存期下降,故miRNA- 21可帮助判断预后[88]。miRNA- 125b在黑色素瘤中表达下降,通过靶向c-jun、MLK3和MKK7抑制肿瘤细胞增殖、周期、凋亡和迁移发挥抑癌基因作用[89],此外还参与了威罗非尼耐药[90]。miRNA- 155在黑色素瘤组织中表达增加,靶向调节SK1,促进凋亡抑制增殖[91]。而miR- 205和miR- 211在黑色素瘤组织中表达下降,分别靶向调节E-cadherin[92]和NFAT3[93]促进肿瘤侵袭。这些miRNA可能作为预测黑色素瘤分期及进展的生物标志物。
2.4 长链非编码RNA
lncRNA是长度大于200个碱基对的非编码RNA。目前,仅有少数lncRNA进行了功能鉴定。根据其编码蛋白基因组的接近程度,lncNA分为正义、反义、内含子、基因间和双向5大类。与miRNA不同,lncRNA可正向或负向调节基因表达,并在lncRNA与RNA、lncRNA与蛋白、lncRNA与染色质间形成许多功能。lncRNA是目前的研究热点,因为大量研究证据表明通过改变lncRNA的原始结构、继发结构和表达水平,将导致从神经退化到癌症的一系列疾病。虽然目前尚无直接证据表明lncRNA是黑色素瘤的生物标志物,但越来越多的研究表明某些lncRNA的异常表达与黑色素瘤患者的生存、侵袭和转移息息相关[94]。例如BANCR与黑色素瘤的生存期相关,其在原发和转移黑色素瘤患者中高表达,并随肿瘤分级增加而表达增加,表明在疾病发展过程中发挥肿瘤基因功能[95- 96],更重要的是,BANCR高表达与低生存期相关,表明BANCR是预后差的生物标志物[97];SPRY4-IT1是移植MAPK通路中的肿瘤抑制因子,其在局部黑色素瘤,远处转移灶淋巴结转移的黑色素瘤中表达增加,预示了其在黑色素瘤分期和早期诊断的潜在功能[98];此外,HOTAIR[99]、 CASC15[100]、 MALAT- 1和 UCA1[101]在黑色素瘤中高表达,可能预示着黑色素瘤的侵袭性和转移性。但lncRNA是否可成为黑色素瘤诊断和预后的生物标志物,尚需进一步研究证实。
3 展望
NGS的普及和表观遗传时代的到来,为探索黑色素瘤生物标志物提供了强有力的工具。随着癌基因组学时代的到来,加之各种先进技术和计算机模型用于大数据分析,联合基因组学、表观遗传组学、蛋白质组学的生物标志物可能辅助黑色素瘤和其他良性肿瘤相鉴别。黑色素瘤容易转移,是致死的重要原因,因此发现器官特异性的生物标志物迫在眉睫。此外,血液中的生物标志物能帮助早期发现黑色素瘤、精准监测疾病进展和预测治疗效果,从而让患者受益。因此,开发新的可用于早期诊断、预测药物敏感性、分析转移可能性和判断预后的标志物,将为黑色素瘤乃至其他恶性肿瘤的治疗和预后提供实验室指导。
[1] Alcala AM, Flaherty KT. BRAF inhibitors for the treatment of metastatic melanoma: clinical trials and mechanisms of resistance[J]. Clin Cancer Res, 2012, 18: 33- 39.
[2] McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM- 3): extended follow-up of a phase 3, randomised, open-label study[J]. Lancet Oncol, 2014, 15: 323- 332.
[3] Queirolo P, Spagnolo F. BRAF plus MEK-targeted drugs: a new standard of treatment for BRAF-mutant advanced melanoma[J]. Cancer Metastasis Rev, 2017,36:35- 42.
[4] Desvignes C, Abirached H, Templier C, et al. BRAF inhibitor discontinuation and rechallenge in advanced mela-noma patients with a complete initial treatment response[J]. Melanoma Res,2017,27:281- 287.
[5] Beaver JA, Theoret MR, Mushti S, et al. FDA approval of nivolumab for the first-lne treatment of patients with BRAFV600 wild-type unresectable or metastatic melanoma[J]. Clin Cancer Res, 2017, 23:3479- 3483.
[6] Meerveld-Eggink A, Rozeman EA, Lalezari F, et al. Short-term CTLA- 4 blockade directly followed by PD- 1 blockade in advanced melanoma patients - a single center experience[J]. Ann Oncol, 2017, 28:862- 867.
[7] Chuk MK, Chang JT, Theoret MR, et al. FDA approval summary: accelerated approval of pembrolizumab for second-line treatment of metastatic melanoma[J]. Clin Cancer Res, 2017,23:5666- 5670.
[8] Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma[J]. Cell,2015, 161: 1681- 1696.
[9] Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma[J]. Nat Genet,2012, 44: 1006- 1014.
[10] Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes[J]. Nature, 2013, 499: 214- 218.
[11] Zhang T, Dutton-Regester K, Brown KM, et al. The genomic landscape of cutaneous melanoma[J]. Pigment Cell Melanoma Res,2016,29: 266- 283.
[12] Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) [J]. Nature,2011,480: 387- 390.
[13] Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma[J]. N Engl J Med, 2012, 367: 107- 114.
[14] Long GV, Weber JS, Infante JR, et al. Overall survival and durable responses in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib combined with trametinib[J]. J Clin Oncol,2016,34: 871- 878.
[15] Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib[J]. N Engl J Med, 2015, 372: 30- 39.
[16] Bahadoran P, Allegra M, Le Duff F, et al. Major clinical response to a BRAF inhibitor in a patient with a BRAF L597R-mutated melanoma[J]. J Clin Oncol,2013, 31: e324- e326.
[17] Marconcini R, Galli L, Antonuzzo A, et al. Metastatic BRAF K601E-mutated melanoma reaches complete response to MEK inhibitor trametinib administered for over 36 months[J]. Exp Hematol Oncol, 2017, 6: 6.
[18] Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study[J]. Lancet Oncol,2013, 14: 249- 256.
[19] Krauthammer M, Kong Y, Bacchiocchi A, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas[J]. Nat Genet, 2015,47: 996- 1002.
[20] Young RJ, Waldeck K, Martin C, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines[J]. Pigment Cell Melanoma Res, 2014, 27: 590- 600.
[21] Diller ML, Kudchadkar RR, Delman KA, et al. Complete response to high-dose IL- 2 and enhanced IFNgamma+Th17: TREG ratio in a melanoma patient[J]. Melanoma Res, 2016,26: 535- 539.
[22] Timar J, Vizkeleti L, Doma V, et al. Genetic progression of malignant melanoma[J]. Cancer Metastasis Rev, 2016, 35: 93- 107.
[23] Li H, Wang Y, Liu H, et al. Genetic variants in the integrin signaling pathway genes predict cutaneous melanoma survival[J]. Int J Cancer, 2017,140: 1270- 1279.
[24] Liang WS, Hendricks W, Kiefer J, et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma[J]. Genome Res,2017, 27: 524- 532.
[25] Royer-Bertrand B, Torsello M, Rimoldi D, et al. Comprehensive genetic landscape of uveal melanoma by whole-genome sequen-cing[J]. Am J Hum Genet, 2016,99: 1190- 1198.
[26] Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation[J]. Nature,2013, 502: 472- 479.
[27] Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET[J]. Science,2009, 324: 930- 935.
[28] Cortellino S, Xu J, Sannai M, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamina-tion-base excision repair[J]. Cell, 2011, 146: 67- 79.
[29] Haffner MC, Chaux A, Meeker AK, et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers[J]. Oncotarget, 2011, 2: 627- 637.
[30] Kim YI, Giuliano A, Hatch KD, et al. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma[J]. Cancer, 1994, 74: 893- 899.
[31] Lee JJ, Murphy GF, Lian CG. Melanoma epigenetics: novel mechanisms, markers, and medicines[J]. Lab Invest,2014,94: 822- 838.
[32] Karpf AR, Matsui S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells[J]. Cancer Res, 2005,65: 8635- 8639.
[33] Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer[J]. Proc Natl Acad Sci USA,1999,96: 8681- 8686.
[34] Sarkar D, Leung EY, Baguley BC, et al. Epigenetic regulation in human melanoma: past and future[J]. Epigenetics, 2015, 10: 103- 121.
[35] De Araujo ES, Kashiwabara AY, Achatz MI, et al. LINE- 1 hypermethylation in peripheral blood of cutaneous melanoma patients is associated with metastasis[J]. Melanoma Res, 2015, 25: 173- 177.
[36] Di JZ, Han XD, Gu WY, et al. Association of hypomethylation of LINE- 1 repetitive element in blood leukocyte DNA with an increased risk of hepatocellular carcinoma[J]. J Zhejiang Univ Sci B,2011,12: 805- 811.
[37] Walesch SK, Richter AM, Helmbold P, et al. Claudin11 promoter hypermethylation is frequent in malignant melanoma of the skin, but uncommon in nevus Cell[J]. Nevi Cancers,2015,7: 1233- 1243.
[38] Cankovic M, Nikiforova MN, Snuderl M, et al. The role of MGMT testing in clinical practice: a report of the association for molecular pathology[J]. J Mol Diagn,2013,15: 539- 555.
[39] Inno A, Fanetti G, Di Bartolomeo M, et al. Role of MGMT as biomarker in colorectal cancer[J]. World J Clin Cases,2014,2: 835- 839.
[40] de Araujo ES, Pramio DT, Kashiwabara AY, et al. DNA methylation levels of melanoma risk genes are associated with clinical characteristics of melanoma patients[J]. Biomed Res Int, 2015, 2015: 376423.
[41] Cheli Y, Ohanna M, Ballotti R, et al. Fifteen-year quest for microphthalmia-associated transcription factor target genes[J]. Pigment Cell Melanoma Res,2010,23: 27- 40.
[42] Ennen M, Keime C, Kobi D, et al. Single-cell gene expression signatures reveal melanoma cell heterogeneity[J]. Oncogene, 2015,34: 3251- 3263.
[43] Hartman ML,Czyz M. MITF in melanoma: mechanisms behind its expression and activity[J]. Cell Mol Life Sci, 2015,72: 1249- 1260.
[44] Mezzanotte JJ, Hill V, Schmidt ML, et al. RASSF6 exhibits promoter hypermethylation in metastatic melanoma and inhibits invasion in melanoma cells[J]. Epigenetics,2014,9: 1496- 1503.
[45] Helmbold P, Richter AM, Walesch S, et al. RASSF10 promoter hypermethylation is frequent in malignant melanoma of the skin but uncommon in nevus cell nevi[J]. J Invest Dermatol, 2012, 132: 687- 694.
[46] Chen H, Zheng Z, Kim KY, et al. Hypermethylation and downregulation of glutathione peroxidase 3 are related to pathogenesis of melanoma[J]. Oncol Rep,2016, 36: 2737- 2744.
[47] Falzone L, Salemi R, Travali S, et al. MMP- 9 overexpression is associated with intragenic hypermethylation of MMP9 gene in melanoma[J]. Aging (Albany NY), 2016, 8: 933- 944.
[48] Gao L, van den Hurk K, Nsengimana J, et al. Prognostic significance of promoter hypermethylation and diminished gene expression of SYNPO2 in melanoma[J]. J Invest Dermatol,2015,135: 2328- 2331.
[49] Muthusamy V, Duraisamy S, Bradbury CM, et al. Epigenetic silencing of novel tumor suppressors in malignant melanoma[J]. Cancer Res, 2006, 66: 11187- 11193.
[50] Curry JL, Richards HW, Huttenbach YT, et al. Different expression patterns of p27 and p57 proteins in benign and malignant melanocytic neoplasms and in cultured human melanocytes[J]. J Cutan Pathol,2009,36: 197- 205.
[51] Liu W, Luo Y, Dunn JH, et al. Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma[J]. J Invest Dermatol,2013, 133: 518- 527.
[52] Koga Y, Pelizzola M, Cheng E, et al. Genome-wide screen of promoter methylation identifies novel markers in melanoma[J]. Genome Res,2009,19: 1462- 1470.
[53] Venza M, Visalli M, Biondo C, et al. Epigenetic marks responsible for cadmium-induced melanoma cell overgrowth[J]. Toxicol In Vitro, 2015, 29: 242- 250.
[54] Furuta J, Umebayashi Y, Miyamoto K, et al. Promoter methy-lation profiling of 30 genes in human malignant melanoma[J]. Cancer Sci,2004, 95: 962- 968.
[55] Liu S, Ren S, Howell P, et al. Identification of novel epigene-tically modified genes in human melanoma via promoter methy-lation gene profiling[J]. Pigment Cell Melanoma Res,2008, 21: 545- 558.
[56] Das AM, Seynhaeve AL, Rens JA, et al. Differential TIMP3 expression affects tumor progression and angiogenesis in melanomas through regulation of directionally persistent endothelial cell migration[J]. Angiogenesis,2014,17: 163- 177.
[57] Schinke C, Mo Y, Yu Y, et al. Aberrant DNA methylation in malignant melanoma[J]. Melanoma Res, 2010, 20: 253- 265.
[58] McGuinness C, Wesley UV. Dipeptidyl peptidase IV (DPPIV),a candidate tumor suppressor gene in melanomas is silenced by promoter methylation[J]. Front Biosci,2008, 13: 2435- 2443.
[59] Matic IZ, Ethordic M, Grozdanic N, et al. Serum activity of DPPIV and its expression on lymphocytes in patients with melanoma and in people with vitiligo[J]. BMC Immunol,2012, 13: 48.
[60] Conway K, Edmiston SN, Khondker ZS, et al. DNA-methylation profiling distinguishes malignant melanomas from benign nevi[J]. Pigment Cell Melanoma Res, 2011,24: 352- 360.
[61] Ekstrom EJ, Sherwood V, Andersson T. Methylation and loss of Secreted Frizzled-Related Protein 3 enhances melanoma cell migration and invasion[J]. PLoS One,2011, 6: e18674.
[62] Tokita T, Maesawa C, Kimura T, et al. Methylation status of the SOCS3 gene in human malignant melanomas[J]. Int J Oncol, 2007, 30: 689- 694.
[63] Fang S, Liu B, Sun Q, et al. Platelet factor 4 inhibits IL- 17/Stat3 pathway via upregulation of SOCS3 expression in melanoma[J]. Inflammation,2014, 37: 1744- 1750.
[64] Bonazzi VF, Nancarrow DJ, Stark MS, et al. Cross-platform array screening identifies COL1A2, THBS1, TNFRSF10D and UCHL1 as genes frequently silenced by methylation in melanoma[J]. PLoS One,2011,6: e26121.
[65] Furuta J, Kaneda A, Umebayashi Y, et al. Silencing of the thrombomodulin gene in human malignant melanoma[J]. Melanoma Res,2005, 15: 15- 20.
[66] Mori T, O’Day SJ, Umetani N, et al. Predictive utility of circulating methylated DNA in serum of melanoma patients receiving biochemotherapy[J]. J Clin Oncol,2005, 23: 9351- 9358.
[67] Lian CG, Xu Y, Ceol C, et al. Loss of 5-hydroxymethylcy-tosine is an epigenetic hallmark of melanoma[J]. Cell,2012,150: 1135- 1146.
[68] Gambichler T, Sand M, Skrygan M. Loss of 5-hydroxymethylcytosine and ten-eleven translocation 2 protein expression in malignant melanoma[J]. Melanoma Res,2013,23: 218- 220.
[69] Lee JJ, Cook M, Mihm MC, et al. Loss of the epigenetic mark, 5-Hydroxymethylcytosine, correlates with small cell/nevoid subpopulations and assists in microstaging of human melanoma[J]. Oncotarget,2015, 6: 37995- 38004.
[70] Lee JJ, Granter SR, Laga AC, et al. 5-Hydroxymethyl-cytosine expression in metastatic melanoma versus nodal nevus in sentinel lymph node biopsies[J]. Mod Pathol,2015, 28: 218- 229.
[71] Pavlova O, Fraitag S, Hohl D. 5-hydroxymethylcytosine expression in proliferative nodules arising within congenital nevi allows differentiation from malignant melanoma[J]. J Invest Dermatol, 2016, 136: 2453- 2461.
[72] Saldanha G, Joshi K, Lawes K, et al. 5-Hydroxymethylcyto-sine is an independent predictor of survival in malignant melanoma[J]. Mod Pathol,2017,30: 60- 68.
[73] Thienpont B, Galle E, Lambrechts D. TET enzymes as oxygen-dependent tumor suppressors: exciting new avenues for cancer management[J]. Epigenomics,2016,8: 1445- 1448.
[74] Gong F, Guo Y, Niu Y, et al. Epigenetic silencing of TET2 and TET3 induces an EMT-like process in melanoma[J]. Oncotarget, 2017,8: 315- 328.
[75] Sigalotti L, Covre A, Fratta E, et al. Epigenetics of human cutaneous melanoma: setting the stage for new therapeutic strategies[J]. J Transl Med,2010, 8: 56.
[76] Ye Y, Jin L, Wilmott JS, et al. PI (4,5)P2 5-phosph-atase A regulates PI3K/Akt signalling and has a tumour suppressive role in human melanoma[J]. Nat Commun, 2013, 4: 1508.
[77] Jazirehi AR,Arle D. Epigenetic regulation of the TRAIL/Apo2L apoptotic pathway by histone deacetylase inhibitors: an attractive approach to bypass melanoma immunotherapy resistance[J]. Am J Clin Exp Immunol, 2013,2: 55- 74.
[78] Zhang XD, Gillespie SK, Borrow JM, et al. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells[J]. Mol Cancer Ther,2004,3: 425- 435.
[79] Bachmann IM, Halvorsen OJ, Collett K, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast[J]. J Clin Oncol,2006,24: 268- 273.
[80] Fan T, Jiang S, Chung N, et al. EZH2-dependent suppres-sion of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression[J]. Mol Cancer Res,2011,9: 418- 429.
[81] Mahmoud F, Shields B, Makhoul I, et al. Role of EZH2 histone methyltrasferase in melanoma progression and metastasis[J]. Cancer Biol Ther, 2016,17: 579- 591.
[82] Sengupta D, Byrum SD, Avaritt NL, et al. Quantitative histone mass spectrometry identifies elevated histone H3 lysine 27 (Lys27) trimethylation in melanoma[J]. Mol Cell Proteomics, 2016, 15: 765- 775.
[83] Ceol CJ, Houvras Y, Jane-Valbuena J, et al. The histone methyltransferase SETDB1 is recurrently amplified in melano-ma and accelerates its onset[J]. Nature, 2011, 471: 513- 517.
[84] Miura S, Maesawa C, Shibazaki M, et al. Immunohistochemistry for histone h3 lysine 9 methyltransferase and demethylase proteins in human melanomas[J]. Am J Dermatopathol, 2014, 36: 211- 216.
[85] Kostaki M, Manona AD, Stavraka I, et al. High-frequency p16(INK) (4A) promoter methylation is associated with histone methyltransferase SETDB1 expression in sporadic cutaneous melanoma[J]. Exp Dermatol, 2014, 23: 332- 338.
[86] Jiang L, Lv X, Li J, et al. The status of microRNA- 21 expression and its clinical significance in human cutaneous malignant melanoma[J]. Acta Histochem, 2012,114: 582- 588.
[87] Zhang J, Lu L, Xiong Y, et al. MLK3 promotes melanoma proliferation and invasion and is a target of microRNA- 125b[J]. Clin Exp Dermatol,2014, 39: 376- 384.
[88] Vergani E, Di Guardo L, Dugo M, et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR- 34a, miR- 100 and miR- 125b[J]. Oncotarget,2016, 7: 4428- 4441.
[89] Levati L, Pagani E, Romani S, et al. MicroRNA- 155 targets the SKI gene in human melanoma cell lines[J]. Pigment Cell Melanoma Res,2011,24: 538- 550.
[90] Liu S, Tetzlaff MT, Liu A, et al. Loss of microRNA- 205 expression is associated with melanoma progression[J]. Lab Invest, 2012,92: 1084- 1096.
[91] Levy C, Khaled M, Iliopoulos D, et al. Intronic miR- 211 assumes the tumor suppressive function of its host gene in melanoma[J]. Mol Cell, 2010, 40: 841- 849.
[92] Rinn JL, Chang HY. Genome regulation by long noncoding RNAs[J]. Annu Rev Biochem,2012, 81: 145- 166.
[93] Wapinski O, Chang HY. Long noncoding RNAs and human disease[J]. Trends Cell Biol, 2011,21: 354- 361.
[94] Li Z, Chao TC, Chang KY, et al. The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL[J]. Proc Natl Acad Sci USA,2014, 111: 1002- 1007.
[95] Guo L, Yao L, and Jiang Y. A novel integrative approach to identify lncRNAs associated with the survival of melanoma patients[J]. Gene,2016,585: 216- 220.
[96] Flockhart RJ, Webster DE, Qu K, et al. BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration[J]. Genome Res,2012, 22: 1006- 1014.
[97] Li R, Zhang L, Jia L, et al. Long non-coding RNA BANCR promotes proliferation in malignant melanoma by regulating MAPK pathway activation[J]. PLoS One, 2014,9: e100893.
[98] Khaitan D, Dinger ME, Mazar J, et al. The melanoma-upre-gulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion[J]. Cancer Res, 2011, 71: 3852- 3862.
[99] Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis[J]. Nature,2010, 464: 1071- 1076.
[100] Lessard L, Liu M, Marzese DM, et al. The CASC15 long intergenic noncoding RNA locus is involved in melanoma progression and phenotype switching[J]. J Invest Dermatol,2015, 135: 2464- 2474.
[101] Tian Y, Zhang X, Hao Y, et al. Potential roles of abnormally expressed long noncoding RNA UCA1 and Malat- 1 in metastasis of melanoma[J]. Melanoma Res,2014,24: 335- 341.
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
昆明医科大学学报(2022年8期)2022-07-31
河北果树(2021年4期)2021-12-02
实用肿瘤学杂志(2020年6期)2020-12-09
福建基础教育研究(2019年10期)2019-05-28
祝您健康·文摘版(2019年1期)2019-05-14
心肺血管病杂志(2018年11期)2018-12-18
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国当代医药(2015年16期)2015-03-01
中国药理学通报(2014年2期)2014-05-09
