
脊髓小脑性共济失调1型一家系谱分析
2018-01-27 01:51刘畅张敏刘恒方陈宁宁崔明吴世陶张植刘勋吴德福徐天阳李明磊
中国神经免疫学和神经病学杂志 2018年1期
刘畅 张敏 刘恒方 陈宁宁 崔明 吴世陶 张植刘勋 吴德福 徐天阳 李明磊
脊髓小脑性共济失调(spinocerebellar ataxias,SCAs)是以进展性的小脑共济失调为表现的常染色体显性遗传性神经系统退行性疾病[1]。SCAs存在多种亚型,且各亚型临床症状相似,交替重叠,通过临床表现进行鉴别诊断困难,只能依靠基因诊断进行分型。其中SCA1型进展比其他常见的SCA2、SCA3、SCA6及SCA7进展更为迅速[2]。目前该病尚无特异性治疗方法,主要为支持疗法,对患者及家庭危害巨大。因此,对有SCA患者的家系成员应早期进行基因诊断,遗传咨询及产前诊断具有重要的临床意义及社会价值。对于没有临床症状的ATXN1基因CAG序列异常扩增者虽不能确诊为SCA1,但可作为遗传咨询。现报告一例SCA1型患者家系谱的分析结果。
1 家系报告
先证者李某,女性,49岁。以“双下肢无力伴步态不稳6年”就诊作者医院神经内科。6年前无明显诱因出现双下肢无力,步态不稳,症状缓慢加重,2年前出现进行性言语不清,音调改变,并出现吞咽困难、进食易呛咳,双上肢不自主震颤,紧张和活动后加重,现已卧床。发病以来,体重下降约10 kg。入院查体:双眼球上视活动不充分,双耳听力下降,伸舌略左偏,舌肌萎缩,舌肌纤颤,四肢肌肉萎缩,左上肢肌力4+级,右上肢肌力5-级,双下肢肌力3级,四肢肌张力正常,四肢腱反射减弱,双侧巴宾斯基征阳性,双侧霍夫曼征阳性,双侧指鼻试验、跟膝胫试验欠稳准。神经电生理检查结果显示,左正中神经、双尺神经、双胫后神经、左腓总神经运动传导速度(MCV)减慢,双正中神经感觉传导速度(SCV)减慢;体感诱发电位(SEP)结果显示双上肢N20未引出波形,双上肢深感觉传导功能异常;脑干听觉诱发电位(BAEP)双侧听性脑干反应(ABR)检测显示,左侧ARB检测发现Ⅰ、Ⅲ、Ⅴ波出波不良,右侧ARB检测发现Ⅲ波潜伏时延迟,波间差延长,Ⅰ、Ⅴ波出波尚可。视觉诱发电位(VEP)检查显示双侧VEP-P100潜伏时未见异常。头颅及脊髓MRI检查见图1。
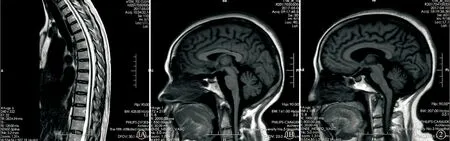
图 1 先证者(Ⅲ7)脊髓MRI检查显示颈胸段脊髓整体偏细(A),头颅MRI检查显示小脑萎缩,未见明显异常信号(B) 图 2 先证者者妹妹(Ⅲ9)头颅MRI检查结果显示小脑萎缩
先证者妹妹(Ⅲ9),女,47岁。6年前无明显诱因出现四肢乏力,步态不稳,双手笨拙,伴言语不清及饮水呛咳,近2年来自觉上诉症状加重。查体:双眼上视不充分,右上肢及双下肢肌张力增高,左上肢肌张力正常,四肢肌力4-级,四肢腱反射正常,双侧巴宾斯基征阳性,双侧指鼻试验、跟膝胫试验欠稳准,闭目难立征阳性。于作者医院行头颅MRI检查结果显示小脑萎缩(图2)。神经电生理检查可见异常(表1)。
先证者第2个妹妹(Ⅲ11),女,44岁。8年前出现双下肢乏力,步态不稳,伴言语不清,饮水呛咳,6年前曾就诊于某医院诊断为“共济失调”,2年前言语不清、步态不稳加重,1年前出现双下肢震颤,紧张及活动后加重。查体:眼球上视时活动不充分,四肢肌张力正常,双上肢肌力5级,双下肢肌力5-级,四肢腱反射正常,双侧巴宾斯基征阳性。
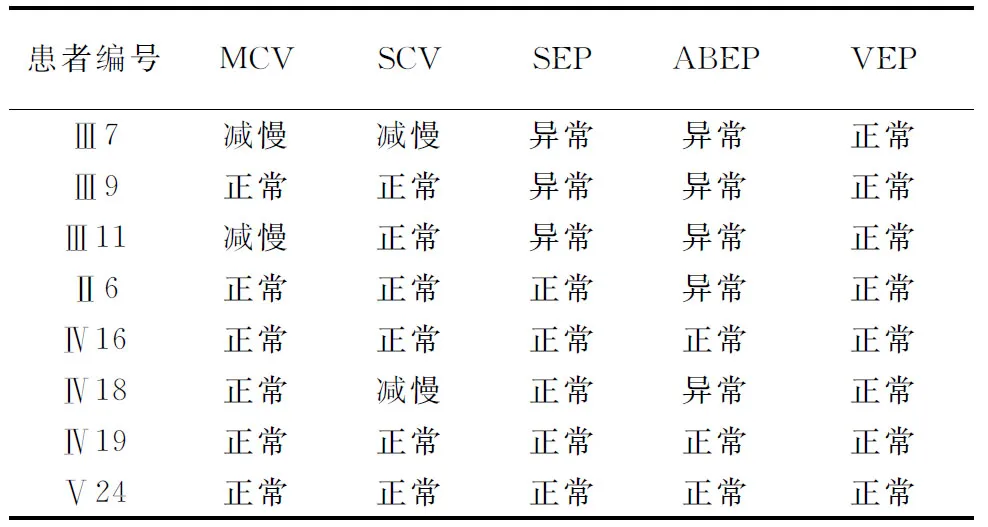
表1 SCA1型患者家系神经电生理检查结果
注:SCA:脊髓小脑性共济失调,表2、图3同;MCV:运动传导速度;SCV:感觉传导速度;SEP:体感诱发电位;BAEP:脑干听觉诱发电位;VEP:视觉诱发电位
先证者姨妈(Ⅱ6),女性,66岁。20年前出现双下肢乏力,站立行走困难,4年前摔倒致左股骨颈骨折,行髋关节置换术后坐轮椅出行。查体:舌肌震颤,四肢肌张力高,双上肢肌力5级,双下肢肌力3级,四肢腱反射正常,双侧指鼻试验、跟膝胫试验欠稳准。
先证者母亲(Ⅱ4)已去世,据家属反映亦有类似临床表现,具体不详。
患者家系中4例为临床表现阳性,均为女性,均排除代谢、中毒等可能影响神经传导的因素,其临床特点见表2。患者家系图谱及神经电生理检查结果见图3和表1。
经患者及家属知情同意,对先证者及家族中4代共8人取血清进行SCAs致病基因检测,包括Ⅱ6、Ⅲ7、Ⅲ9、Ⅲ11、Ⅳ16(30岁)、Ⅳ18(22岁)、Ⅳ19(9岁)、Ⅴ24(5岁)。血液标本送至北京信诺佰世医学检验所采用PCR-STR分析法检查SCAs相关基因,结果显示8人的SCA1致病基因ATXN1基因CAG均发生异常扩增(正常参考范围为<39次),其中家系Ⅲ11CAG重复次数为45次,Ⅱ6重复次数为39次,余6名成员(Ⅲ7、Ⅲ9、Ⅳ16、Ⅳ18、Ⅳ19、Ⅴ24)重复次数均为44次。
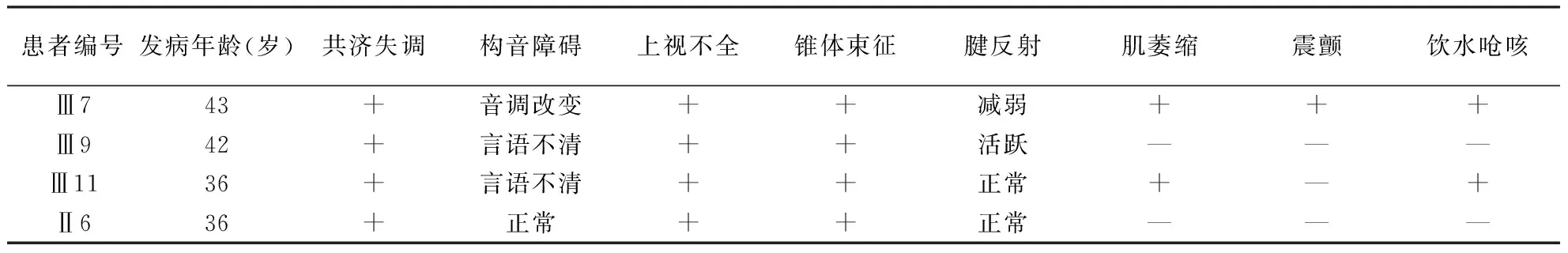
表2 SCA1家系中4例有症状者临床表现
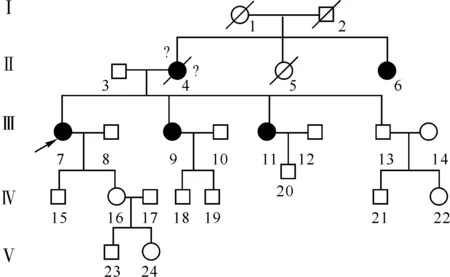
注::键康男性;:健康女性;男性患者;:女性患者;:先证者;:死亡;?:疑似患者 图 3 SCA1患者家系图谱
2 讨论
流行病学研究表明,SCA1患者的患病率为(1~2)/10万,世界上SCA1患者占所有SCA类型的6%[1]。其临床表现复杂,主要为进展性的小脑共济失调及脑干功能损害,早期可表现为步态失调、搅拌式语言、平衡困难、眼球震颤以及吞咽困难,后期表现为眼球慢扫视运动、眼球上视不充分、辨距不良、轮替动作障碍、肌张力减弱,进展期可出现肌萎缩、本体感觉缺失、认知损害、舞蹈症、张力失常、延髓功能损害症状[1]、声带麻痹。本家系4例患者的发病情况基本符合SCA1型特点,均以步态、肢体的共济失调为首发症状,Ⅱ6的症状最轻,仅表现为肢体共济失调及眼球上视不充分,Ⅲ7、Ⅲ9和Ⅲ11在此表现上还出现构音障碍,先证者症状最重伴有音调改变,很难辨别其说话的内容,先证者Ⅲ7和Ⅲ11有舌肌萎缩及舌肌纤颤、饮水呛咳,四肢肌肉萎缩无法自行行走,并且伴有肢体震颤,基本与文献报道的SCA1型复杂的症状相符合,难以严格按照临床表现分期,并且本家系出现的肢体震颤文献中尚未提及,在Ⅱ代及Ⅲ代观察到了遗传早现现象。
SCA是常染色体显性遗传病,有20余种基因型,不同分型其临床特点以及预后各不相同。SCA2、SCA3和SCA1最难鉴别,其临床特点相似,交替重叠[3-4]。SCA2患者主要表现为身体上半部分的共济失调,以及明显、频繁的姿势性震颤,眼慢扫视运动突出,腱反射消失严重;SCA3患者在疾病早期可出现明显的锥体外系症状,有时伴有轻微的共济失调,眼球震颤、眼球上视不充分症状比SCA1患者出现的几率更大且更早[4];另外有研究证实,SCA1比SCA2及SCA3在1年内进展更为迅速,故当患者在小脑共济失调基础上出现脑干功能损害,可首先进行基因检测这几型[1]。严重并且广泛的腱反射消失多见于SCA2、SCA3及SCA4型,但SCA1型不常见[5]。SCA6发病较晚,通常在50~60岁发病。SCA17的症状更为多样,并伴随锥体外系症状及认知水平下降。SCA5、SCA6及SCA8通常表现为单纯的小脑症状,很少有表明广泛的神经系统病变的症状,进展速度较SCA1慢。如患者出现黄斑病变引起的视力下降,几乎可以把SCA7的诊断放到第一位,并且家族里有视力下降的表现也可以考虑SCA7[1,6]。
神经电生理研究发现,SCA1患者一般有神经传导异常,41%的患者有异常的VEP,41% SEP异常,73% BAEP异常,运动诱发电位在大部分患者中也存在异常[7-8]。此家系中对8名成员进行了神经电生理检查,均显示有不同程度的异常,在临床症状为SCA1的4例患者尤为典型,其中MCV 异常2例,SCV异常1例,SEP异常3例,BEAP异常4例,VEP均为正常,考虑SCA1主要累及小脑及脑干,对于患者的运动、浅感觉、深感觉及听觉诱发都有不同程度的影响,此4例SCA1患者的临床表现及病理表现与之相符。由于该研究病例数较少,有关SCA1的具体电生理表现有待于进一步研究。
已有症状SCA1患者经磁共振形态测量学(voxel-based morphometry)检查可发现脑干、小脑灰质及白质体积减少,亦可见脊髓萎缩[1]。家系Ⅲ9患者MRI结果显示小脑萎缩,颈胸段脊髓整体偏细。SCA1患者出现临床症状之前,常规MRI对这个阶段的敏感性较低,Quantitative MRI可记录到轻微的运动神经元变性,小脑萎缩以及脑干灰质萎缩[9-10]。PET(positron emission tomography)可检测到小脑、脑桥甚至小脑幕上的低代谢,表明这些结构发生神经元变性[11]。对于家系里无临床症状者可行这两项检查,以了解脑部情况。
SCA1 是位于常染色体6p22-23上的ATXN1基因CAG序列异常扩增,在编码ataxia-1蛋白时产生异常的多聚谷氨酰胺链延长,干扰正常的蛋白功能,导致神经毒性[1,3]。ATXN1基因CAG序列异常扩增达39或以上时可提示SCA1[1]。但基因学检测对于无临床症状的发现CAG异常扩增的高危人群只能作为基因咨询,尚不能直接确诊,所以该家族里经检测存在ANTX1的CAG序列有异常扩增但无临床症状的另4人暂不能诊断为SCA1。SCA1一般于30~40岁发病,外显率超过95%,与年龄相关,但在儿童时期以及60岁以后发病者也偶有记录[12]。欧洲一项对317例确诊为SCA1的研究表明,ANTX1基因上扩增的和正常的序列均对于预测发病年龄起到很大的作用[13],SCA1患者的发病年龄受ATXN1扩增链和正常链共同影响,而且正常链发挥积极的影响,所以CAG序列扩增的次数也不能用于预测被检者的发病年龄、临床表现以及进展速度,可一定程度上作为参考[1,13]。有证据表明CAG的重复次数与疾病严重程度密切相关,重复次数越多,疾病发生的年龄越早并且越严重[2,13]。该家系患者Ⅲ11经基因检测CAG序列重复次数最多,其发病年龄为36岁,是家族中最早发病者,且进展程度相对Ⅲ9及Ⅱ6迅速;Ⅱ6是CAG序列重复最少者,且临床症状也最轻。成年后发病的SCA1患者,从开始发病至死亡时间间隔一般为10~30年;儿童时期发病的SCA1(13岁前症状出现)一般病情进展更迅速,症状更严重,一般于16岁前夭折[1]。研究结果显示,于婴儿和青少年时期发病的SCA1者可检测出存在大量重复的CAG序列,一般达50次以上,患者病程进展迅速且大部分为男性,多有面肌舌肌萎缩、构音困难、骨骼肌萎缩、眼肌麻痹表现,且与疾病进程不相关[1]。该家系中尽管Ⅳ19和Ⅴ24为年幼的扩增基因携带者,但未检测到存在大量重复的CAG序列,推测其发生严重的进展迅速的青少年SCA1概率较低,应密切跟踪随访其30岁以后的情况。
综上所述,SCA1患者的临床表现丰富多样,虽然具有一定特点,但不能依靠临床表现及MRI检查对SCA1和其他类型的SCA进行鉴别诊断,其确诊必须依赖分子基因学检测。SCA患者后代发病率较高,且SCA1进展最为迅速,对家族成员尽早进行基因筛查对家庭及社会具有重要意义。
[1]Opal P,Ashizawa T. Spinocerebellar ataxia type 1[M]. In:Adam MP,Ardinger HH,Pagon RA,et al. GeneReviews® [Internet]. Seattle (WA):University of Washington,Seattle.1993—2017.
[2]Ashizawa T,Figueroa KP,Perlman SL,et al. Clinical characteristics of patients with spinocerebellar ataxias 1,2,3 and 6 in the US:a prospective observational study[J]. Orphanet J Rare Dis,2013,8:177.
[3]俞李强,何晓辉,方琪,等.脊髓小脑性共济失调症状前诊断初探[J].中国神经免疫学和神经病学杂志,2013,20(3):197-199.
[4]Jacobi H,Reetz K,du Montcel ST,et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1,2,3,and 6 in the longitudinal RISCA study:analysis of baseline data[J]. Lancet Neurol,2013,12:650-658.
[5]Ragno M,Perretti AC,Castaldo I,et al. Multimodal electrophysiologic follow-up study in 3 mutated but presymptomatic members of a spinocerebellar ataxia type 1 (SCA1) family[J].Neurol Sci,2005,26:67-71.
[6]Emir UE,Brent Clark H,Vollmers ML,et al. Non-invasive detection of neurochemical changes prior to overt pathology in a mouse model of spinocerebellar ataxia type 1[J]. J Neurochem,2013,127:660-668.
[7]Chandran V,Jhunjhunwala K,Purushottam M,et al. Multimodal evoked potentials in spinocerebellar ataxia types 1,2,and 3[J]. Ann Indian Acad Neurol,2014,17:321-324.
[8]Schmitz-Hübsch T,Fimmers R,Rakowicz M,et al. Responsiveness of different rating instruments in spinocerebellar ataxia patients[J]. Neurology,2010,74:678-684.
[9]Jacobi H,du Montcel ST,Bauer P,et al. Long-term disease progression in spinocerebellar ataxia types 1,2,3,and 6:a longitudinal cohort study[J]. Lancet Neurol,2015,14:1101-1108.
[10]Pedroso JL,de Souza PV,Pinto WB,et al. SCA1 patients may present as hereditary spastic paraplegia and must be included in spastic-ataxias group[J]. Parkinsonism Relat Disord,2015,21:1243-1246.
[11]Miyai I,Ito M,Hattori N,et al. Cerebellar ataxia rehabilitation trialists collaboration. Cerebellar ataxia rehabilitation trial in degenerative cerebellar diseases[J]. Neurorehabil Neural Repair,2012,26:515-522.
[12]朱杨帆,陈涛,杨丹,等.遗传性脊髓小脑共济失调3型MRI检查的应用及进展[J]. 中国神经免疫学和神经病学杂志,2015,22(2):130-132.
[13]Tezenas du Montcel S,Durr A,Rakowicz M,et al. Prediction of the age at onset in spinocerebellar ataxia type 1,2,3 and 6[J].Med Genet,2014,51:479-486.
猜你喜欢
中国医药导报(2022年28期)2022-11-25
中国现代医生(2022年21期)2022-08-22
临床输血与检验(2022年3期)2022-06-22
当代医药论丛(2021年24期)2022-01-20
山东医药(2021年24期)2021-09-01
诊断学(理论与实践)(2020年1期)2020-04-28
中国医学影像技术(2020年11期)2020-01-13
创新作文(小学版)(2019年4期)2019-07-24
郑州大学学报(医学版)(2019年3期)2019-06-03
中国医药科学(2017年9期)2017-08-04
