
X连锁鱼鳞病并发Meleda角化病一例临床特征及SLURP⁃1和STS基因突变分析
2017-12-28 17:30王艳汪慧君林志淼胡凌寒潘玉雪刘晓雁杨勇
中华皮肤科杂志 2017年11期
王艳 汪慧君 林志淼 胡凌寒 潘玉雪 刘晓雁 杨勇
100034北京大学第一医院皮肤科[王艳(现在山西医科大学第二医院皮肤科,030001太原)、汪慧君、林志淼、胡凌寒、潘玉雪、杨勇];首都儿科研究所附属儿童医院皮肤科(刘晓雁)
·论著·
X连锁鱼鳞病并发Meleda角化病一例临床特征及SLURP⁃1和STS基因突变分析
王艳 汪慧君 林志淼 胡凌寒 潘玉雪 刘晓雁 杨勇
100034北京大学第一医院皮肤科[王艳(现在山西医科大学第二医院皮肤科,030001太原)、汪慧君、林志淼、胡凌寒、潘玉雪、杨勇];首都儿科研究所附属儿童医院皮肤科(刘晓雁)
目的报道1例X连锁鱼鳞病并发Meleda角化病,并检测其基因突变。方法收集临床资料,提取患儿及其父母外周血基因组DNA,PCR扩增SLURP⁃1和STS基因全部外显子及其侧翼序列,以100例健康人作为对照,对扩增产物行琼脂糖凝胶电泳检测,并对SLURP⁃1基因扩增产物进行DNA测序。结果患儿躯干、四肢泛发规则排列的棕褐色或黑色多角形鳞屑,掌跖、肘膝、腹股沟、肛周红斑,过度角化,向背侧延伸,诊断为X连锁鱼鳞病并发Meleda角化病。基因检测提示,STS全基因缺失;SLURP⁃1基因第3外显子第286位核苷酸发生C→T纯合突变(c.286C>T),导致其编码蛋白质在第96位氨基酸出现终止改变(p.R96*),其父母均为c.286C>T杂合突变携带者。健康对照未发现此突变。结论该患者携带STS全基因缺失和SLURP⁃1基因纯合无义突变,可能是导致X连锁鱼鳞病并发Meleda角化病的原因。
鳞癣,X染色体连锁;皮肤角化病,掌跖;突变;基因缺失;基因,STS;基因,SLURP⁃1
鱼鳞病是一种常见的角化障碍性皮肤病,其中X连锁鱼鳞病(X⁃linked ichthyosis,XLI)发病率约1/6 000,属于常见的遗传性鱼鳞病,一般只发生于男性,女性为携带者[1]。患者出生时或生后不久即发病,表现为全身皮肤干燥、粗糙,覆黑褐色鳞片,主要累及肢体伸侧,也可累及屈侧,颈部及耳前区受累为该病特征。Meleda角化病(mal de Meleda)是一种罕见的掌跖角化病(palmoplantar keratoderma,PPK),属于常染色体隐性遗传,表现为弥漫性掌跖增厚,累及手足背侧及肘膝。我们报道首例XLI合并Meleda角化病的患儿,并对其进行致病基因检测。
资料与方法
一、病历资料
患儿男,4岁,出生时即有全身皮肤干燥,伴有脱屑,手足皮肤正常,生后5~6个月时,双手掌、足底出现红斑、皮肤增厚,逐渐加重,发展至手足背部,伴有多汗。2岁时肘膝出现红色角化性斑块,并累及双侧腹股沟、臀部等部位,无自觉不适。患儿无鱼鳞病及掌跖角化病家族史。父母体健,为同一镇人,否认近亲结婚,患儿有2姐,均体健。体检:各系统检查无异常。皮肤科检查:全身皮肤干燥,头皮、颈部、躯干、四肢泛发棕褐色或黑色多角形、黏着性鳞屑,排列规则,腹部较背部为重;双手掌、足底融合性角质增生性斑块,呈蜡样黄色,上有脱屑及皲裂,向手足背部延伸,边缘发红,双侧肘部、腹股沟、阴囊、肛周、双膝伸侧、腘窝、双踝散发大小不等境界清楚的红色角化性斑块,手指皮肤增厚角化,可见收缩带,指趾间散在白色浸渍,手足活动正常,功能不受限(图1、2)。根据患儿颈部、躯干和四肢出现大而黑的鱼鳞样黏着性鳞屑,腹部受累较背部更多,呈进行性加重,手、足、肘、膝角化性斑块,伴有越线现象,临床诊断为XLI并发Meleda角化病。经北京大学医学信息咨询中心检索证明,国内外检索暂未发现与本研究内容相同的文献报道。患儿外用10%尿素乳膏后,皮肤干燥、脱屑及手、足、肘、膝角化明显减轻。
二、方法
1.外周血基因组DNA提取:本研究通过北京大学第一医院医学伦理委员会批准,患儿监护人签署知情同意书。取患儿、父母和100例健康对照外周血4 ml,2%乙二胺四乙酸抗凝,用试剂盒提取基因组DNA(北京天根生化科技有限公司)。
2.PCR引物设计和合成:根据SLURP⁃1、STS基因序列(http://www.ensembl.org/index.html),用Primer⁃BLAST 在 线 版(https://www.ncbi.nlm.nih.gov/tools/primer⁃blast/)SLURP⁃1设计3对引物(表1),STS设计10对特异性引物(表2),覆盖上述2条基因的所有外显子及侧翼序列,引物由北京天一辉远生物科技有限公司合成。

图1 患儿X连锁鱼鳞病皮损表现 1A:腹部、上肢多角形鳞屑;1B:双下肢伸侧黏着性黑色鳞屑;1C:双下肢屈侧皮肤干燥,泛发黑色鳞屑

图2 患儿Meleda角化病皮损 2A:手掌黄色角化性斑块、皲裂、脱屑,弥漫至手腕部,边缘发红;2B:足背红斑;2C:腹股沟、阴囊红斑;2D:双膝伸侧角化性斑块
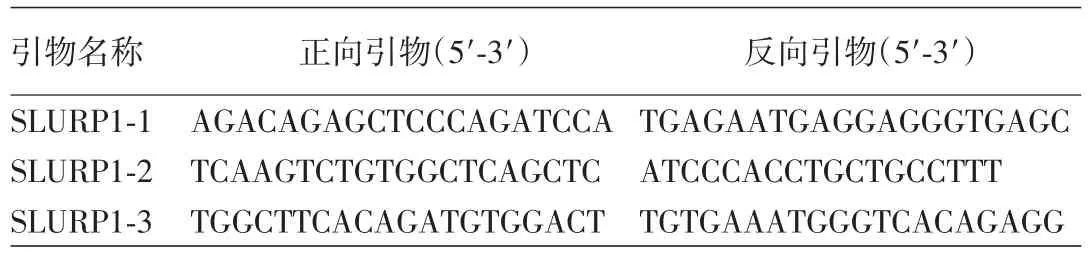
表1 SLURP1引物序列
3.PCR反应体系及条件:扩增用法国Transgene公司的2倍EasyTaq PCR SuperMix(批号AS111⁃11),反应体系25 μl。反应条件:94℃预变性5 min,94℃变性30 s,62℃退火30 s(每个循环依次降0.6℃),72℃延伸30 s,10个循环;94℃变性 30 s,57℃退火30 s,72℃延伸30s,25个循环;72℃延伸10 min,4℃保存。取5 μl PCR扩增产物行10 g/L琼脂糖凝胶电泳检测。
4.PCR产物测序分析:SLURP⁃1 PCR产物纯化后送至北京天一辉远生物科技有限公司测序,测序方法为Sanger双脱氧链终止法。测序结果采用Bioedit软件与SLURP⁃1基因组序列进行比对。

表2 STS引物序列
结 果
一、STS基因PCR扩增结果
见图3。琼脂糖凝胶电泳显示,10对引物所扩增的阳性对照(健康人)均得到对应条带,患儿及阴性对照(ddH2O)无条带出现,表明该患儿STS全基因缺失。
二、测序结果
将SLURP⁃1所测序列与Ensembl数据库(http://ensembl.org/index.html)公布的正常序列进行比对,结果示SLURP⁃1基因第3外显子中第286位核苷酸(从cDNA编码起始位点ATG算起)发生C→T纯合突变(c.286C>T)(dbSNP:rs121908317),导致其编码蛋白质在第96位氨基酸出现无义突变(p.R96*)(图4A)。健康对照未出现此突变(图4C),父母经检测为c.286C>T杂合突变携带者(图4B)。所有突变均由反向测序得到验证。

图3 STS基因扩增产物琼脂糖凝胶电泳图 M:标准参照物;1~10:外显子1~10;p:患儿DNA扩增产物;+:健康人阳性对照;-:阴性对照
讨 论
XLI为X染色体连锁隐性遗传,患者几乎均为男性,没有明显的种族或地域差异。90%的XLI患者在生后第1周即出现全身皮肤干燥、脱屑,颈部、躯干、四肢逐渐出现大而黑的多角形黏着性鳞屑,屈侧受累明显,手足皮肤正常,症状不随年龄增长而减轻,皮肤外表现包括角膜混浊、睾丸肿瘤、隐睾等。寻常性鱼鳞病患者身体屈侧及颈部不受累,常伴掌跖皮纹增多及毛周角化症,可与XLI鉴别。本文患儿为男性,出生即有全身皮肤干燥,随年龄增长进行性加重,出现颈部、腹部及四肢大而黑的多角形鳞屑,腹部受累较背部明显,且向下延伸至小腿伸侧,符合XLI临床表现。

图4 SLURP⁃1基因突变测序图 4A:患儿SLURP⁃1第3外显子第286位核苷酸发生纯合突变(C.286C>T);4B:患儿父亲为C.286C>T杂合突变携带者;4C:健康人测序结果
1978年,Webster等[2]发现,XLI与皮肤缺乏类固醇硫酸酯酶(steroid sulfatase,STS)有关;1987年,STS基因缺失被确定为XLI的病因[3]。正常情况下,STS主要存在于角质层细胞膜中,此酶的主要功能是将角质层中的胆固醇硫酸盐水解,胆固醇硫酸盐已证明在保持膜的完整性以及维持角质层的正常脱落中起重要作用[4]。当STS缺乏时,角质层的胆固醇硫酸盐不能被分解,出现鳞屑堆积;另一方面胆固醇硫酸盐分解过程被阻断,皮肤的屏障作用也受到干扰,pH值下降,角质层黏附性增加,进一步使角质层细胞滞留,促进鳞屑堆积,从而出现相应的临床表现[5]。文献表明,90%的XLI患者STS全基因缺失[6⁃7]。我们采用扩增STS 10对外显子的方法确定患儿为STS全基因缺失,与文献报道一致,结合病史、临床表现及基因检测结果,进一步明确XLI的诊断。2010年西班牙研究者对40例XLI患者进行STS基因检测,发现部分缺失率达25%,而基因缺失程度与表型之间未发现相关性[6]。
Meleda角化病是一个罕见的常染色体隐性遗传性掌跖角化病,在1826年由Luca Stulli在亚得里亚海的Meleda岛首次发现,1898年才由Neumann首次报道[8],其患病率约为1/10万[9]。临床表现有口周红斑、跨关节部位皮肤的角化过度性斑块、指甲畸形、浸渍多汗伴有恶臭、指过短、假指等[10]。长岛型掌跖角化病也为早期出现的掌跖部位弥漫性红斑及角化,伴有越线现象,但皮疹稳定,不随年龄增长出现明显进展或加重。Olmsted综合征口周红斑更严重,并伴有残肢现象。该患儿出生后5~6个月时出现掌跖角化,并逐渐累及手背、足背、阴囊、肛周、腹股沟及腘窝等部位,伴有多汗、浸渍,无残肢现象,根据临床表现诊断为Meleda角化病。
2001年,Fischer等[11]确认Meleda角化病为编码SLURP⁃1蛋白的基因突变所致。这类蛋白是分泌Ly⁃6/uPar蛋白超家族中的一员。SLURP⁃1 3D结构含有3个结构域,5个双硫键,这对其正确折叠行使功能至关重要。迄今为止,全世界不同人群中已检测到16个SLURP⁃1基因的致病突变位点[12]。本文患儿SLURP⁃1第3外显子中第286位核苷酸C→T发生纯合无义突变(c.286C>T),该突变在Fischer等关于Meleda角化病患者基因突变研究的文献中曾被报道[11],亦证实该患儿Meleda角化病诊断。SLURP⁃1被认为是晚期上皮分化的标志,参与调节角质形成细胞的生长、增殖、分化和凋亡[13],广泛分布于人体皮肤、宫颈、牙龈、胃和食管,尤其在掌跖的角质形成细胞中含量最高。SLURP⁃1是α⁃7烟碱乙酰胆碱受体的内源性配体,具有促凋亡活性[14]。当SLURP⁃1蛋白失活时,由于角质形成细胞凋亡不能被正常调控,导致皮肤高度角化。SLURP⁃1还有抑制巨噬细胞和角质形成细胞释放肿瘤坏死因子α的功能。Meleda角化病患者缺乏SLURP⁃1蛋白会导致肿瘤坏死因子α的释放及随后的炎症介质不能被正常调控[15],从而引起持续炎症,这和很多肿瘤形成有关,包括恶性黑素瘤。既往认为Meleda角化病除在亚得里亚海的Meleda岛有较高发病率外,中东和地中海地区也有,在这些地区近亲结婚较常见,提示祖先效应[10]。截至目前已有包括阿尔及利亚、土耳其、巴勒斯坦、巴基斯坦、中国台湾、德国、苏格兰、日本、利比亚等多达10余个国家的病例报道[16⁃17]。2016年我国Zhang等[12]首次报道两个通过基因检测确诊为Meleda角化病的家系,3例患者均发生c.256G>A错义突变。
部分鱼鳞病可以伴有掌跖角化,但是XLI和Meleda角化病具有不同的遗传方式,同时出现于同一个患者,国内外尚未见报道,其具体机制不明。该患儿无鱼鳞病及掌跖角化病家族史,同胞两姐均正常,父母亲否认近亲结婚,但是均携带相同的SLURP⁃1基因杂合突变,考虑原因为双方均出生在人口仅为1 000余人的同一小镇,可能存在较近的血缘关系。目前鱼鳞病与掌跖角化病治疗主要是口服维A酸类药物,或者外用角质剥脱剂、润肤剂等。本例患儿XLI和Meleda角化病临床表现均较严重,可能与SLURP⁃1基因突变和STS基因缺失发生在同一患儿从而产生协同效应有关。
[1]Nagtzaam IF,Stegmann AP,Steijlen PM,et al.Clinically manifest X⁃linked recessive ichthyosis in a female due to a homozygous interstitial 1.6⁃Mb deletion of Xp22.31[J].Br J Dermatol,2012,166(4):905⁃907.DOI:10.1111/j.1365⁃2133.2011.10685.x.
[2]Webster D,France JT,Shapiro LJ,et al.X⁃linked ichthyosis due to steroid⁃sulphatase de fi ciency[J].Lancet,1978,1(8055):7072.
[3]Bonifas JM,Morley BJ,Oakey RE,et al.Cloning of a cDNA for steroid sulfatase:frequent occurrence of gene deletions in patients with recessive X chromosome ⁃linked ichthyosis[J].Proc Natl Acad Sci U S A,1987,84(24):9248⁃9251.
[4]Wang N,An K,Liu H,et al.Detection of the STS gene in a family with X⁃linked recessive ichthyosis[J].Indian J Dermatol Venereol Leprol,2013,79(2):268.DOI:10.4103/0378⁃6323.107669.
[5]Elias PM,Williams ML,Choi EH,et al.Role of cholesterol sulfate in epidermal structure and function:lessons from X⁃linked ichthyosis[J].Biochim Biophys Acta,2014,1841(3):353⁃361.DOI:10.1016/j.bbalip.2013.11.009.
[6]Cañueto J,Ciria S,Hernández⁃Martín A,et al.Analysis of the STS gene in 40 patients with recessive X⁃linked ichthyosis:a high frequency of partial deletions in a Spanish population[J].J Eur Acad Dermatol Venereol,2010,24(10):1226 ⁃1229.DOI:10.1111/j.1468⁃3083.2010.03612.x.
[7]Takeichi T,Sugiura K,Hsu CK,et al.Novel indel mutation of STS underlies a new phenotype of self⁃healing recessive X⁃linked ichthyosis[J].J Dermatol Sci,2015,79(3):317 ⁃319.DOI:10.1016/j.jdermsci.2015.07.001.
[8]Taylor JA,Bondavalli D,Monif M,et al.Mal de Meleda in Indonesia:mutations in the SLURP1 gene appear to be ubiquitous[J].Australas J Dermatol,2016,57(1):e11⁃13.DOI:10.1111/ajd.12239.
[9]Bchetnia M,Laroussi N,Youssef M,et al.Particular Mal de Meleda phenotypes in Tunisia and mutations founder effect in the Mediterranean region[J/OL].Biomed Res Int,2013,2013:206803[2017 ⁃05 ⁃25].http://dx.doi.org/10.1155/2013/206803.DOI:10.1155/2013/206803.
[10]Gruber R,Hennies HC,Romani N,et al.A novel homozygous missense mutation in SLURP1 causing Mal de Meleda with an atypical phenotype[J].Arch Dermatol,2011,147(6):748⁃750.DOI:10.1001/archdermatol.2011.138.
[11]Fischer J,Bouadjar B,Heilig R,et al.Mutations in the gene encoding SLURP⁃1 in mal de Meleda[J].Hum Mol Genet,2001,10(8):875⁃880.
[12]Zhang J,Cheng R,Ni C,et al.First Mal de Meleda report in Chinese Mainland:two families with a recurrent homozygous missense mutation in SLURP ⁃1[J].J Eur Acad Dermatol Venereol,2016,30(5):871⁃873.DOI:10.1111/jdv.13038.
[13]Morais eSFA,Cunha TV,Boeno ES,et al.Mal de Meleda:a report of two cases of familial occurrence[J].An Bras Dermatol,2011,86(4 Suppl 1):S100⁃103.
[14]Vilas⁃Sueiro A,Rosón E,Suárez⁃Peñaranda JM,et al.Malignant melanoma in a patient with mal de Meleda[J].Clin Exp Dermatol,2016,41(4):437⁃439.DOI:10.1111/ced.12791.
[15]Perez C,Khachemoune A.Mal de Meleda:a focused review[J].Am J Clin Dermatol,2016,17(1):63⁃70.DOI:10.1007/s40257⁃015⁃0157⁃1.
[16]Sakabe J,Kabashima⁃Kubo R,Kubo A,et al.A Japanese case of Mal de Meleda with SLURP1 mutation[J].J Dermatol,2014,41(8):764⁃765.DOI:10.1111/1346⁃8138.12539.
[17]Bchetnia M,Bozgia M,Laroussi N,et al.The first Mal de Meleda case in Libya:identification of a SLURP1 mutation[J].Int J Dermatol,2015,54(12):1426⁃1428.DOI:10.1111/ijd.12373.
A case of X⁃linked ichthyosis complicated by Mal de Meleda:clinical features and mutation analysis of the SLURP⁃1 and STS genes
Wang Yan,Wang Huijun,Lin Zhimiao,Hu Linghan,Pan Yuxue,Liu Xiaoyan,Yang Yong
Department of Dermatology,Peking University First Hospital,Beijing 100034,China(Wang Y[current affiliation:Department of Dermatology,The Second Hospital of Shanxi Medical University,Taiyuan 030001,China],Wang HJ,Lin ZM,Hu LH,Pan YX,Yang Y);Department of Dermatology,Children′s Hospital,Capital Institute of Pediatrics,Beijing 100020,China(Liu XY)
Yang Yong,Email:dryongyang@bjmu.edu.cn
ObjectiveTo report a case of X⁃linked ichthyosis complicated by Mal de Meleda,and to identify the gene mutations.MethodsClinical data were collected from the patient,and peripheral blood samples were obtained from the patient,his parents and 100 unrelated healthy people who served as controls.Genomic DNA was extracted from these blood samples,and PCR was performed to amplify all the exons and their flanking sequences of the SLURP⁃1 and STS genes.All the amplification products were analyzed by agarose gel electrophoresis,and amplification products of the SLURP⁃1 gene were analyzed by DNA sequencing.ResultsThe patient presented with regularly⁃arranged polygonal brown or black scales all over the trunk and limbs,erythematous hyperkeratotic lesions on the palms and soles,elbows and knees,inguinal and perianal regions,which extended to the dorsa of the hands and feet.Then,the patient was diagnosed with X⁃linked ichthyosis complicated by Mal de Meleda.Genetic testing showed complete deletion of the STS gene,and a homozygous mutation(c.286C > T)at position 286 in exon 3 of the SLURP⁃1 gene,which led to the formation of a premature termination codon at amino acid position 96(p.R96*).His parents were heterozygous carriers of the mutation(c.286C > T).No mutation was found in the unrelated healthy controls.ConclusionThe complete deletion of the STS gene and the homozygous nonsense mutation in the SLURP⁃1 gene may be the reason for X⁃linked ichthyosis complicated by Mal de Meleda in the patient.
Ichthyosis,X ⁃linked;Keratoderma,palmoplantar;Mutation;Gene deletion;Gene,STS;Gene,SLURP⁃1
杨勇,Email:dryongyang@bjmu.edu.cn
10.3760/cma.j.issn.0412⁃4030.2017.11.008
2017⁃06⁃22)
吴晓初)
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国美容医学(2022年2期)2022-03-17
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
家庭医药(2018年2期)2018-02-09
东方教育(2017年14期)2017-09-25
家庭医药(2016年4期)2016-05-04
课程教育研究·学法教法研究(2016年1期)2016-03-17
