
黏质沙雷氏菌plaA及plaS的生物信息学分析
2017-12-19 10:49:12王青青丁恒武刘必融薛正莲阚显照
生物学杂志 2017年6期
王 莹, 王 洲, 章 勤, 王青青, 蒋 澜, 吴 璇, 丁恒武, 刘必融, 薛正莲, 阚显照
(1. 安徽师范大学 生命科学学院 生物信息研究所, 芜湖 241000; 2. 安徽工程大学 生物与化学工程学院, 芜湖 241000)
黏质沙雷氏菌plaA及plaS的生物信息学分析
王 莹1,2, 王 洲2, 章 勤1, 王青青1, 蒋 澜1, 吴 璇1, 丁恒武1, 刘必融1, 薛正莲2, 阚显照1
(1. 安徽师范大学 生命科学学院 生物信息研究所, 芜湖 241000; 2. 安徽工程大学 生物与化学工程学院, 芜湖 241000)
磷脂酶A1是一类水解磷脂的酶类,利用生物信息学方法对磷脂酶A1基因及辅助蛋白基因进行分析,为进一步研究磷脂酶提供基础。通过对黏质沙雷氏菌plaA(Phospholipase A1)及plaS(accessory protein) 进行克隆和测序,对其核苷酸序列和氨基酸序列特征进行分析;对其蛋白质二级结构、三级结构进行了预测;同时运用ML、MP和BI 3种方法构建了系统发育树。结果表明,plaS的dN/dS的值较plaA高,所受的选择压力较小,进化速率较快;PlaA蛋白和PlaS蛋白的二级结构都以α螺旋和无规则卷曲为主;plaA与plaS联合构建的系统发育树支持率较16S单独构建的支持率高。基于plaA、plaS和 16S 3个基因联合构建的系统发育树中,解脲沙雷氏菌和嗜线虫沙雷氏菌都与黏质沙雷氏菌聚为一支,在亲缘关系上,这两种沙雷氏菌可能跟黏质沙雷氏菌更为接近。
磷脂酶A1基因;辅助蛋白基因plaS;沙雷氏菌;系统发育
磷脂酶A1是一类水解磷脂sn-1位酰基的酶类[1],广泛存在于自然界,在动物[2-4]、植物[3,5]和微生物中均有发现。含有磷脂酶A1的微生物主要包括耶和森菌[6]、微白黄链霉菌[7]、曲霉菌[8]、假单胞杆菌[9]和沙雷氏菌[10-12]等。磷脂酶A1主要应用于食品、制药和生物燃料产业[13]。磷脂酶A1在工业上得到广泛应用,目前研究材料多来自于沙雷氏菌属。
Song等[14]将沙雷氏菌MK1磷脂酶A1基因(plaA)克隆到大肠杆菌中,发现其下游的plaS也参与该基因的表达;plaA编码321氨基酸,与液化沙雷氏菌(Serratialiquefaciens)和结肠炎耶尔森杆菌(Yersiniaenterocolitica)的plaA核苷酸序列相似度达到70%左右。付建红[15]、苏燕南[16]等发现plaS编码一段为224个氨基酸的蛋白,该蛋白没有酶活性,可能对于磷脂酶A1基因的表达有辅助作用。
目前,磷脂酶A1的研究主要是在工业上,而对于plaA及辅助蛋白基因plaS的生物信息学方面研究较少。沙雷氏属间的系统发育关系并不是很清楚,且多是利用单基因16S进行研究探讨,利用单基因进行研究的不足之处在于构建的系统发育树支持率较低。所以本研究对我们自主分离的黏质沙雷氏菌ESE-2014的plaA和plaS进行扩增、克隆和测序,并利用生物信息学方法,主要探讨:1)plaA和plaS的序列结构特征;2)PlaA蛋白的理化性质及沙雷氏属间的进化速率;3)产磷脂酶的沙雷氏菌属之间的系统发育关系。从而为进一步探讨磷脂酶的功能以及提高磷脂酶活性提供基础。
1 材料和方法
1.1 材料
1.1.1 菌株的来源
黏质沙雷氏菌菌株(ESE-2014)由安徽工程大学微生物发酵安徽省工程技术研究中心分离和培养。
1.2 方法
1.2.1 黏质沙雷氏菌基因组DNA的提取
所选实验材料总DNA的提取采用传统的酚/氯仿抽提法[17]。
1.2.2 磷脂酶A1基因的克隆
根据NCBI公布的磷脂酶A1基因序列(No.JX138535)及含有辅助蛋白的磷脂酶A1基因plaS序列(No.JX138537)设计引物(表1)分别扩增磷脂酶A1基因plaA和含有辅助蛋白的磷脂酶A1基因plaS。以黏质沙雷氏菌ESE-2014基因组为模板,进行PCR扩增,将PCR扩增得到的目的片段进行克隆和回收并测序。引物由通用生物系统(安徽)有限公司合成。
PCR扩增采用25 μL反应体系:10×PCR buffer 2.5 μL, dNTPs (2 mmol/L) 3 μL, primer-F(5 μmol/L) 2.2 μL, primer-R (5 μmol/L) 2.2 μL, TaKaRaTaq(5 U/μL) 0.13 μL, template 1 μL, ddH2O 13.97 μL。PCR反应体系为:70℃预变性4 min;94℃变性40 s,53℃退火20 s,72℃延伸2 min 30 s, 共4个循环; 94℃变性20 s,53℃退火20 s, 72℃延伸2 min 30 s, 共36个循环; 最后再72℃延伸10 min。
PCR扩增结束后进行琼脂糖凝胶电泳检测。PCR产物经纯化克隆选取阳性克隆,克隆试剂盒为p-EASY-T5 Zero Cloning Kit。挑取的阳性克隆由通用生物系统(安徽)有限公司进行测序。
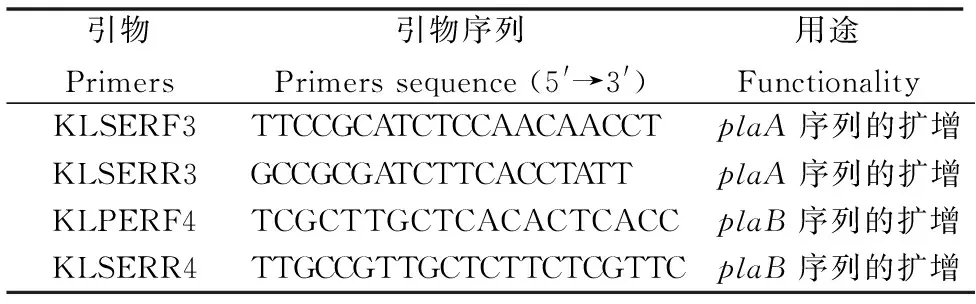
表1 本研究所用的引物
1.2.3 基因的核苷酸序列分析及碱基替代模型分析
运用BioEdit 7.2.5分析plaA和plaS的AT含量;使用ModelGenerator V. 0.851软件对核苷酸替代最适模型进行分析,通过Bayesian Information Criterion(BIC)统计后估算。
1.2.4 磷脂酶A1蛋白分析
蛋白质的理化性质主要利用ExPASy在线软件分析(http://web.expasy.org/protparam/);二级结构的预测则是利用在线网站NPS@SOMPA进行分析(https://npsa-prabi.ibcp.fr);利用TMHMM 2.0实现跨膜区结构的预测(http://www.cbs.dtu.dk/services/TMHMM-2.0/);蛋白质三级结构采用PHD[18]软件在线分析平台预测(http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index)。
1.2.5 进化速率分析
基于磷脂酶A1基因plaA和含有辅助蛋白的磷脂酶A1基因plaS序列,对本文测序的序列并联合GenBank中已释放的其他10个种50条序列进行选择压力分析,所受的选择压力分析使用Datamonkey(http://www.datamonkey.org)[19]在线分析平台,分别对plaA、plaS的平均dN/dS进行计算。
1.2.6 系统发育树的构建
建树前使用MAFFT软件进行序列比对。
最大简约法 (MP) 分析,使用PAUP*4.0b10软件。采用启发式 (heuristic) 搜索最大简约树 (MP),序列添加方式选用100次的随机分类群重复,树等分与重连分支交换法 (TBR)获取系统树。自展法(bootstrap)重复检验1000次,用以分析系统发育树拓扑结构的可靠性。
最大似然法(ML)分析,使用RaxML GUI v.1.3.1软件。核苷酸替代模型采用GTRCAT,ML + slow bootstrap,run 10次,通过1000次重复的自展法评估ML树上分支的可靠性。
贝叶斯法(BI)分析,使用软件MrBayes 3.2.3软件构建贝叶斯树,氨基酸和核苷酸的序列分段计算模型,起始树设为随机树,3条热链和一条冷链共4条马尔可夫链(Markov chains),运算10 000 000代(每隔1000代抽样1次),前25%的树舍弃,余下的75%的树推测贝叶斯后验概率和支持率高于50%的一致树。马尔可夫链都运算2次,这样可以保证得到可信的贝叶斯后验概率。
2 结果与分析
2.1 plaA及plaS的扩增和序列分析
利用两对引物KLSERF3/ KLSERR3、KLPERF4/KLSERR4分别对黏质沙雷氏菌plaA、plaS进行PCR扩增,获得963 bp、756 bp两条序列。将所得的序列在NCBI中进行BLAST比对,结果显示,plaA与黏质沙雷氏菌PL-06plaA(No.JX138535)相似度达到99%,plaS与黏质沙雷氏菌PL-06的plaS(No.JX138537)相似度达到99%,由此推测,扩增获得的目的片段为plaA、plaS序列。
沙雷氏菌plaA长度为963到969 bp,AT含量为33.64%~47.25%(表2)。以黏质沙雷氏菌ESE-2014为例,基因长度为963 bp,AT含量为35.83%。plaS长度在705到772 bp,AT含量为30.15%~46.83%。小肠结肠炎耶尔森杆菌和鼠疫耶尔森氏菌(Yersiniapestis)plaA、plaS序列AT%含量最高,plaA序列AT含量分别为50.97%~50.36%,plaS序列AT含量分别为52.51%~51.60%。黏质沙雷氏菌AT含量在34%~38%左右,菌株FGI94 AT为38.42%;普城沙雷氏菌(Serratiaplymuthica)plaA序列AT含量在39%~41%,plaS序列AT含量在38%左右。在沙雷氏菌属中,其中居泉沙雷氏菌(Serratiafonticola)plaA、plaS序列AT含量最高,无花果沙雷氏菌(Serratiaficaria)plaA、plaS序列AT含量最低,分别为33.64%~30.15%。
表2 本研究物种的来源及plaA和plaS的GenBank登录号
(续表2 Continued Table 2)
菌株StrainsplaA登录号GenBankaccessionnumberplaS登录号GenBankaccessionnumber液化沙雷氏菌SerratialiquefaciensHUMV-21CP011303CP011303液化沙雷氏菌SerratialiquefaciensATCC27592NC_021741NC_021741液化沙雷氏菌SerratialiquefaciensFDAARGOS_125CP014017CP014017普城沙雷氏菌SerratiaplymuthicaAS9NC_015567NC_015567普城沙雷氏菌SerratiaplymuthicaV4CP007439CP007439普城沙雷氏菌Serratiaplymuthica3Rp8CP012096CP012096普城沙雷氏菌Serratiaplymuthica3Re4-18CP012097CP012097普城沙雷氏菌SerratiaplymuthicaS13NC_021659NC_021659普城沙雷氏菌SerratiaplymuthicaRVH1NZ_ARWD01000001NZ_ARWD01000001普城沙雷氏菌Serratiaplymuthica4Rx13NC_021591NC_021591普城沙雷氏菌SerratiaplymuthicaA153LRQU01000001LRQU01000001居泉沙雷氏菌SerratiafonticolaGS2CP013913CP013913居泉沙雷氏菌SerratiafonticolaDSM4576CP011254CP011254格氏沙雷氏菌SerratiagrimesiiA2JGVP01000019JGVP01000019无花果沙雷氏菌SerratiaficariaNBRC102596BCTS01000010BCTS01000010深红沙雷氏菌Serratiarubidaea1122CP014474CP014474小肠结肠炎耶尔森杆菌Yersiniaenterocolitica8081CP009846CP009846鼠疫耶尔森氏菌YersiniapestisCO92CP009973CP009973

图1 PlaA蛋白序列比对
Fig 1 PlaA protein sequence alignment
2.2 磷脂酶A1蛋白质分析
2.2.1 磷脂酶A1蛋白基本信息的获取及理化特性分析
通过同源比对(相似度高于75%),利用NCBI网站获取沙雷氏菌磷脂酶A1蛋白序列,利用ExPASy在线软件分析它们的理化性质。PlaA蛋白分子质量大约在28.9~33.7 ku,等电点为4.71~5.58,其中居泉沙雷氏菌蛋白分子质量和等电点较高,居泉沙雷氏菌菌株GS2等电点最高。
2.2.2 PlaA蛋白序列比对分析
对沙雷氏属10个种的PlaA蛋白序列进行了比对分析(图1)。沙雷氏属的PlaA蛋白序列长度为318~320个氨基酸, 序列相对比较保守,有多个保守区域,分别在45~54、56~74、91~99、195~211、214~224、244~258的位置。其中居泉沙雷氏菌和深红沙雷氏菌在8~55的位置变异比较大。
2.2.3 磷脂酶A1蛋白二级结构预测及跨膜区结构分析
对于黏质沙雷氏菌菌株ESE-2014的PlaA和PlaS蛋白,利用在线网站NPS@SOMPA预测其蛋白质的二级结构。黏质沙雷氏菌ESE-2014 PlaA和PlaS蛋白二级结构都主要是以α螺旋和无规则卷曲为主,PlaA蛋白α螺旋和无规则卷曲所占的比例分别为38.75%和35.94%,PlaS蛋白α螺旋占的比例高达58.57%,无规则卷曲所占的比例为35.94%。通过TMHMM分析表明,PlaA蛋白没有跨膜结构,PlaS蛋白则有一个跨膜区,所在的位置为7~26。
2.2.4 磷脂酶A1蛋白三级结构预测
蛋白质的三级结构主要预测方法包括同源建模、折叠识别和从头预测,由于已知蛋白的序列与结构数据库中模板蛋白的序列一致性小于30%,所以采用折叠识别法预测PlaA和PlaS蛋白的三维结构。利用phyre2在线网站预测结果显示,折叠子d3tgla与PlaA蛋白的匹配性非常高,三级结构主要由数个α螺旋和β片组成,PlaS蛋白则是无规则卷曲连接的多个α螺旋构成,与折叠子c4cj9A匹配性非常高(图2)。
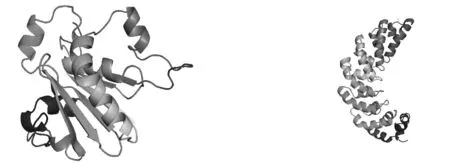
图2 黏质沙雷氏菌ESE-2014 PlaA/PlaS蛋白三级结构预测图
注:图左为PlaA蛋白三级结构, 图右为PlaS蛋白质三级结构
2.2.5 沙雷氏菌的进化速率分析
每个基因的进化速率都不相同,同一个基因的不同序列位置进化速率也不同,因此,在进行系统进化之前对基因的进化速率进行评估比较相当重要。本文通过对plaA及plaS在沙雷氏属的非同义替换(dN)和同义替换(dS)之间的比值进行计算,分别从种上和种下水平来探讨plaA和plaS的进化特征。在遗传学中,dN/dS表示的是非同义替换(dN)和同义替换(dS)之间的比值。核苷酸位点可以根据密码子的简并性(degeneracy)分成两类:同义替换位点(Synonymous site)和非同义替换位点(Non-synonymous site),同义替换是指引起所编码的氨基酸发生的替换是不改变,同义替换受到的选择作用比较小;非同义替换是指所编码的氨基酸发生改变的替换。如果dN/dS>1,则认为基因受正选择(positive selection),dN/dS=1,则基因受中性进化(neutral evolution),dN/dS<1时,则基因受纯化选择(purify selection)。在黏质沙雷氏菌种下水平上,黏质沙雷氏菌plaA的dN/dS为0.105,plaS为0.158;在普城沙雷氏菌种下水平上,普城沙雷氏菌plaA的dN/dS为0.124,plaS为0.288;沙雷氏属plaA的dN/dS为0.135,plaS为0.202。在沙雷氏菌种下水平上,黏质沙雷氏菌、普城沙雷氏菌plaS的dN/dS值较高;在沙雷氏属间,也是plaS的dN/dS值较高,plaS的所受的选择压力相对较小,进化速率相对较快。
2.2.6 系统发育分析
经过Mafft软件序列比对,我们得到了用于系统发育分析的数据矩阵: 1)plaA序列长度为1005 bp。2)plaS序列长度为775 bp。3)16S序列长度为1545 bp。4)plaA和plaS整合序列组长度为1780 bp。5)plaA、plaS和16S整合序列组长度为3308 bp。用Bayesian Information Criterion(BIC)法确定plaA序列的最佳模型为TrN+G;plaS为K81uf+G,16S序列的最佳模型为HKY+I。系统发育树图的枝长和拓扑结构来自最大似然图。鼠疫耶尔森氏菌(Yersiniapestis)和小肠结肠炎耶尔森菌(Yersiniaenterocolitica)作为外类群,其余物种作为内类群。节点上的数字按顺序分别表示ML、MP的bootstrap支持率及BI法的后验概率。用MP、ML和BI 3种方法对数据组进行分析,均得到相同或相似的拓扑结构图,不同之处是节点的支持率(或后验概率)及枝长。
基于plaA、plaS联合构建的系统发育树中,主要分成六大枝,各分支的支持率和后验概率较高,同一个物种不同菌株聚在一起。基于plaA、plaS和16S联合构建的系统发育树中,对沙雷氏属10个物种的系统发育关系进行探讨,ML和MP法构建的系统发育树拓扑结构一致,支持率较高。
3 讨论
基于plaA、plaS构建的系统发育树(图3),同一个物种的不同菌株大都聚在一起,黏质沙雷氏菌聚为一大支,再和无花果沙雷氏菌聚为一支,居泉沙雷氏菌单独聚为一支位于基部。Bhadra等[20]认为解脲沙雷氏菌NiVa51T利用尿素作为氮源,DNA杂交及生理学和生物学上的实验表明该菌株基因型和表型都不同于沙雷氏菌,提出NiVa51T作为一个新种;Zhang等[21]同样提出嗜线虫沙雷氏菌DZ0503SB1T是一个新种。基于磷脂酶基因构建的系统发育树中,解脲沙雷氏菌Lr5/4 LG59与黏质沙雷氏菌RSC-14互为姐妹类群,ML和MP的bootstrap值分别为88和90,BI后验概率为1.00;嗜线虫沙雷氏菌DZ0503SB1与黏质沙雷氏菌B3R3聚为一小支,ML和MP的bootstrap值分别为70和62,BI后验概率为0.93,然后和黏质沙雷氏菌U36365聚为一支,支持率和后验概率都非常高,ML和MP支持率都为100,BI后验概率为1.00;解脲沙雷氏菌和嗜线虫沙雷氏菌都聚在黏质沙雷氏菌这一大支上,从系统发育树上来看,解脲沙雷氏菌和嗜线虫沙雷氏菌与黏质沙雷菌亲缘关系更为接近。
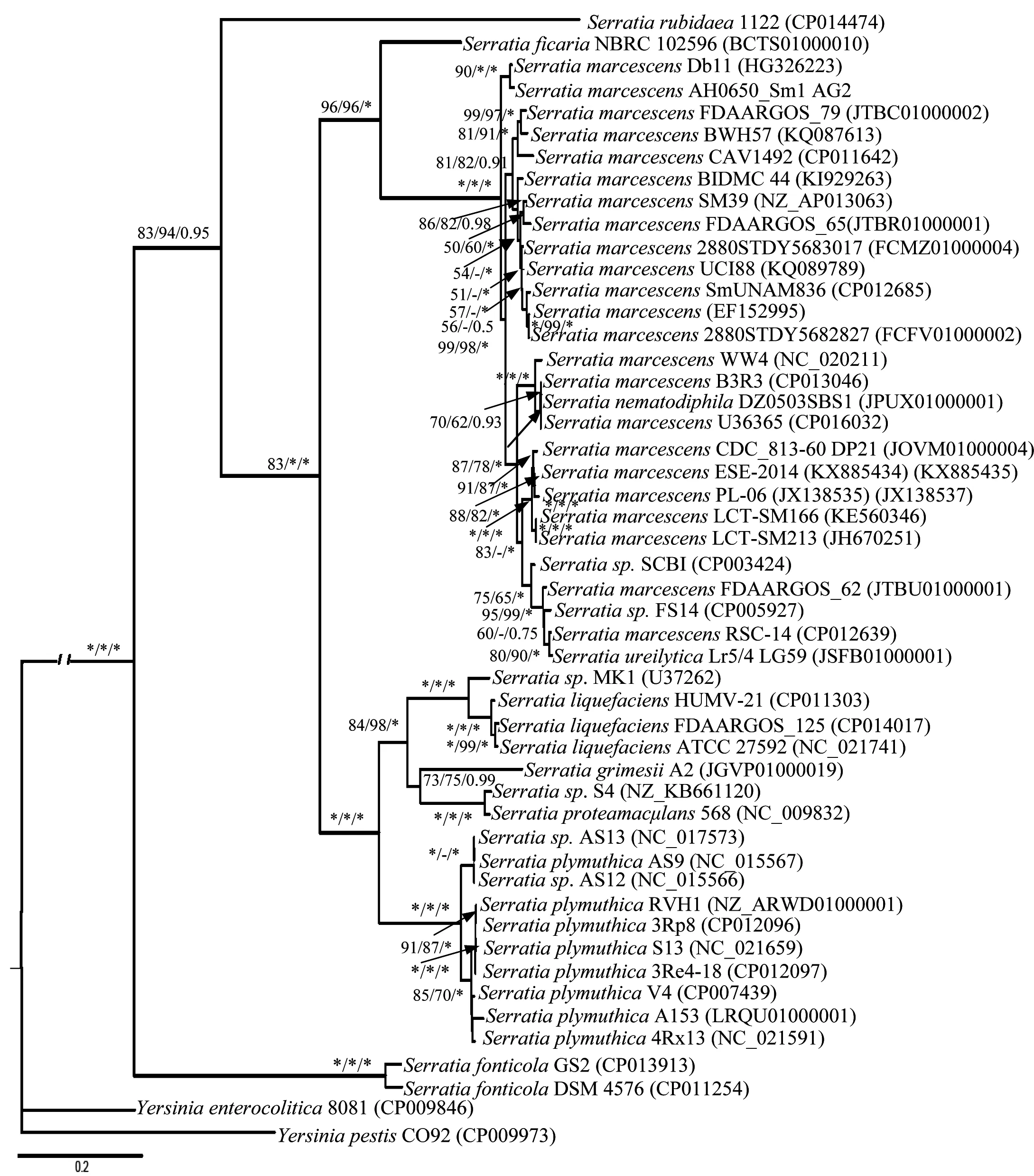
图3基于plaA、plaS的核苷酸序列运用ML、MP和BI法构建的系统发育树(各节点上的数字指的分别是ML、MP支持率和贝叶斯后验概率)
Fig 3 Phylogenetic tree based on the nucleotide dataset ofplaA,plaSgenes using ML, MP and BI methods. Numbers by nodes refer to ML, MP bootstrap values and Bayesian posterior probability
注:系统发育树的拓扑结构图采用ML法的树图;“*”表示ML、MP支持率为100(%)或贝叶斯后验概率为1.00;“-”表示ML、MP支持率低于50%或后验概率小于0.5
基于plaA、plaS和16S构建基因树(图4),使用了最大简约法(MP)、最大似然法(ML)和贝叶斯法(BI),其中ML法和MP法构建的系统发育树拓扑结构一致,BI法存在微小差异。黏质沙雷氏菌和解脲沙雷氏菌聚为一支,再和嗜线虫沙雷氏菌聚在一起;居泉沙雷氏菌单独聚为一支,ML和MP支持率都为100%,BI后验概率为1。García Fraile[22]利用16S构建的ML树中有大都一致的拓扑结构,但是大都非常低,说明仅用16S构建系统发育树来探讨沙雷氏属间的关系不够充分。而基于磷脂酶基因和16S联合构建的系统发育树,ML、MP和BI法的支持率和后验概率都较16S单独构树高,能够反映沙雷氏菌种间的发育关系。
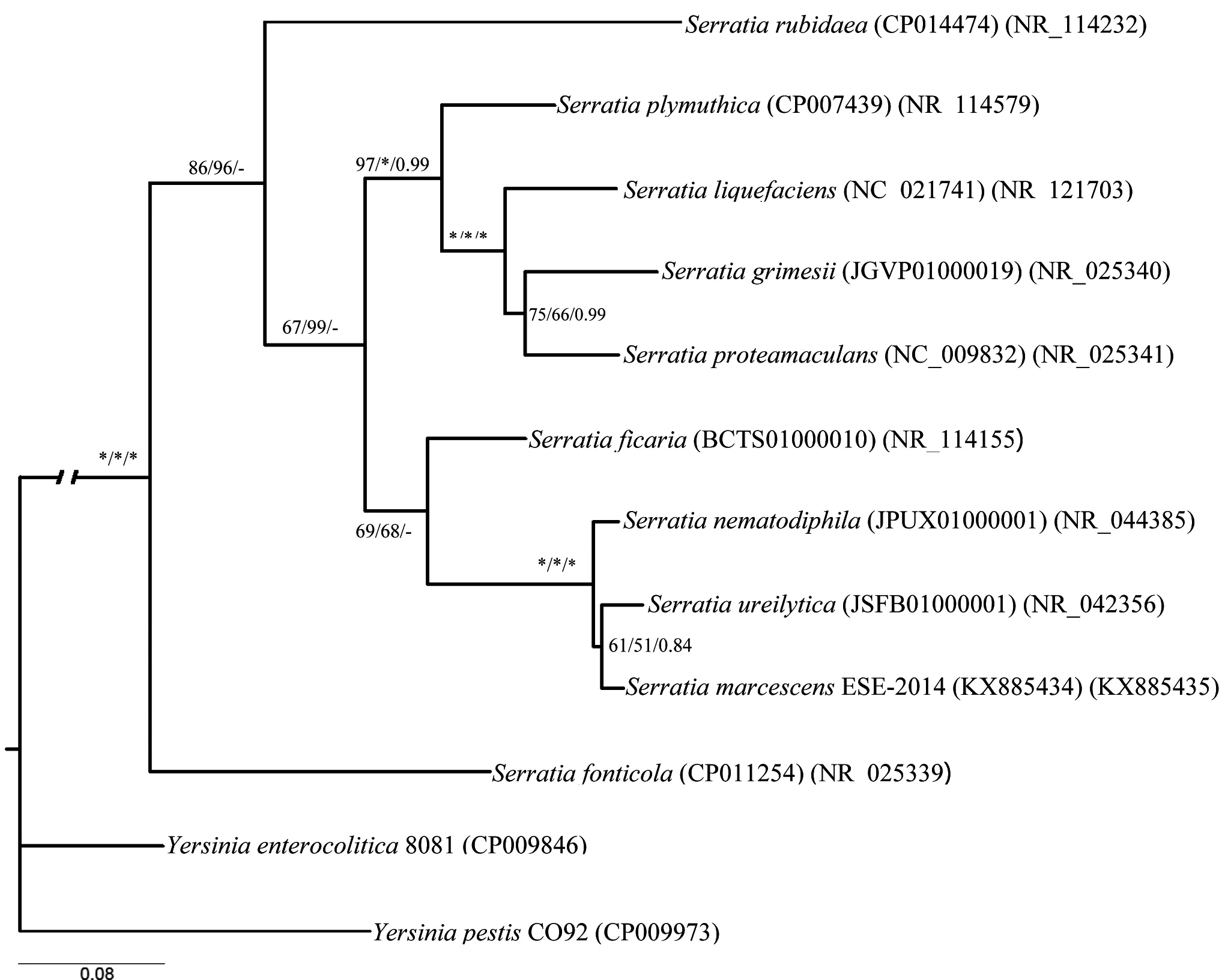
图4基于plaA、plaS和16S核苷酸序列的沙雷氏属系统发育树(各节点上的数字指的分别是ML、MP支持率和贝叶斯后验概率)
Fig 4 Phylogenetic tree based on the nucleotide dataset ofplaA,plaSand 16S using ML, MP and BI methods ofSerratia. Numbers by nodes refer to ML, MP bootstrap values and Bayesian posterior probability
注:系统发育树的拓扑结构图采用ML法的树图;“*”表示ML、MP支持率为100(%)或贝叶斯后验概率为1.00;“-”表示ML、MP支持率低于50%或后验概率小于0.5
[1]MOHARANA T R, BYREDDY A R, PURI M, et al. Selective enrichment of omega-3 fatty acids in oils by phospholipase A1[J]. PLoS One, 2016, 11(3):e0151370.
[2]RICHMOND G S, SMITH T K. A novel phospholipase fromTrypanosomabrucei[J]. Molecular Microbiology, 2007, 63(4):1078-1095.
[3]RICHMOND G S, SMITH T K. The role and characterization of phospholipase A1 in mediating lysophosphatidylcholine synthesis inTrypanosomabrucei[J]. Biochemical Journal, 2007, 405(2):319-329.
[4]PEREZ-RIVEROL A, PEREIRA F D, MUSACCHIO LASA A, et al. Molecular cloning, expression and IgE-immunoreactivity of phospholipase A1, a major allergen fromPolybiapaulista(Hymenoptera: Vespidae) venom[J]. Toxicon, 2016, 124:44-52.
[5]ISHIGUROS, KAWAI-ODA A,UEDA J, et al. 拟南芥花药不开裂基因(DAD1)编码一个新的磷脂酶A1、其催化茉莉酸生物合成的最初步骤,并与花粉成熟,花药开裂和开花同步进行[J]. 植物学通报,2002,19(3):384.
[6]BAKHOLDINA S I, TISCHENKO N M, SIDORIN E V, et al. Recombinant phospholipase A1 of the outer membrane of psychrotrophicYersiniapseudotuberculosis: expression, purification, and characterization[J]. Biochemistry, 2016, 81(1):47-57.
[7]SAKASEGAWA S I, MAEBA R, MURAYAMA K, et al. Hydrolysis of plasmalogen by phospholipase A1 fromStreptomycesalbidoflavusfor early detection of dementia and arteriosclerosis[J]. Biotechnology Letters, 2016, 38(1):109-116.
[8]HAMA S, ONODERA K, YOSHIDA A, et al. Improved production of phospholipase A 1 by recombinantAspergillusoryzaethrough immobilization to control the fungal morphology under nutrient-limited conditions[J]. Biochemical Engineering Journal, 2015, 96:1-6.
[9]NISHIHARA M, KAMATA M, KOYAMA T, et al. New phospholipase A1-producing bacteria from a marine fish[J]. Marine Biotechnology, 2008, 10(4):382-387.
[10]GIVSKOV M, MOLIN S. Secretion ofserratialiquefaciensphospholipase fromEscherichiacoli[J]. Molecular Microbiology, 1993, 8(2):229-242.
[11]KIM M K, KIM J K, RHEE D J S. Isolation of a phospholipase A 1-producing microorganism[J]. Journal of Industrial Microbiology, 1996, 16(3):171-174.
[12]付建红, 唐辉桂, 姚 斌, 等. 一株产低温碱性磷脂酶A1耐冷细菌的筛选及发酵条件的初步研究[J]. 工业微生物 2008, 38(5):12-16.
[13]LIM H J, PARK Y J, JANG Y J, et al. Cell-free synthesis of functional phospholipase A1 fromSerratiasp.[J]. Biotechnology for Biofuels, 2016, 9:159.
[14]SONG J K, KIM M K, RHEE J S. Cloning and expression of the gene encoding phospholipase A 1 fromSerratiasp . MK1 inEscherichiacoli[J]. Journal of Biotechnology, 1999, 72(12):103-114.
[15]FU J, HUANG H, MENG K, et al. A novel cold-adapted phospholipase A 1 fromSerratiasp. xjF1: Gene cloning, expression and characterization[J]. Enzyme & Microbial Technology, 2008, 42(2):187-194.
[16]苏燕南, 薛正莲, 陈 涛, 等. 粘质沙雷氏菌PL-06磷脂酶A1基因大肠杆菌优化表达[J]. 中国生物工程杂志, 2013, 33(7):36-42.
[17]SAMBROOK J F, RUSSELL D W. Molecular cloning: a laboratory manual (3-Volume Set) [M]. New York: Cold Spring Harbor Laboratory Press, 2001.
[18]KELLEY L A, STERNBERG M J. Protein structure prediction on the web: a case study using the Phyre server[J]. Nature Protocol, 2009, 4(3):363-371.
[19]DELPORT W, POON A F, FROST S D, et al. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology[J]. Bioinformatics, 2010, 26(19):2455-2457.
[20]BHADRA B, ROY P, CHAKRABORTY R.Serratiaureilyticasp. nov., a novel urea-utilizing species[J]. International Journal of Systematic & Evolutionary Microbiology, 2005, 55(Pt5):2155-2158.
[21]ZHANG C X, YANG S Y, XU M X, et al.Serratianematodiphilasp. nov., associated symbiotically with the entomopathogenic nematodeHeterorhabditidoideschongmingensis(Rhabditida: Rhabditidae) [J]. International Journal of Systematic & Evolutionary Microbiology, 2009, 59(Pt7):1603-1608.
BioinformaticsanalysisofplaAandplaSinSerratiamarcescens
WANG Ying1, 2, WANG Zhou2, ZHANG Qin1, WANG Qing-qing1, JIANG Lan1, WU Xuan1, DING Heng-wu1, LIU Bi-rong1, XUE Zheng-lian2, KAN Xian-zhao1
(1. The Institute of Bioinformatics, College of Life Sciences, Anhui Normal University, Wuhu 241000; 2. College of Biological and Chemical Engineering, Anhui Engineering University, Wuhu 241000, China)
Phospholipase A1 (plaA) is an enzyme that hydrolyzes phospholipids. We used bioinformatics methods to analyzeplaAand its accessory protein gene (plaS), which will provide a basis for further study of phospholipase. We cloned and sequenced theplaAandplaSofSerratiamarcescens, and the characteristics of nucleotide and amino acid sequence were analysed. Furthermore, we also predicted the secondary structure and tertiary structure of PlaA and PlaS protein. Phylogenetic trees were reconstructed using maximum parsimony (MP), maximum likelihood (ML) and bayesian inference (BI) methods. The results showed that the dN / dS ofplaSwas higher than that ofplaA, suggesting that the selection pressure is stronger and the evolution rate is slower. The secondary structure of PlaA and PlaS protein were mainly composed of α-helix and random coil. Bootstrap values of 16S tree were lower than that of trees referred from two genes (plaA+plaS). Based on the phylogenetic tree which constructed byplaA,plaSand 16S, bothSerratianematodiphilaandS.ureilyticawere clustered together withS.marcescens. The two strains ofSerratiamay be more related toS.marcescens.
phospholipase A1; accessory protein geneplaS;Serratia; phylogeny
2016-11-08;
2016-11-17
国家自然科学基金(NO.31471615)
王 莹,硕士研究生,研究方向为微生物基因组学, E-mail:wangyingad@163.com
薛正莲,教授,研究方向为工业微生物育种及发酵过程优化,E-mail: xuezhen0851@sina.com;阚显照,教授,博士生导师,研究方向为分子系统与进化,E-mail: xianzhao@ahnu.edu.cn
10.3969/j.issn.2095-1736.2017.06.001
Q951.3
A
2095-1736(2017)06-0001-06
猜你喜欢
温州大学学报(自然科学版)(2022年2期)2022-05-30 14:03:48
环境污染与防治(2022年5期)2022-05-29 08:35:24
昆明医科大学学报(2021年2期)2021-03-29 07:42:16
工程数学学报(2020年3期)2020-07-06 07:38:40
长治学院学报(2019年2期)2019-07-24 07:14:04
食品与发酵工业(2018年11期)2018-12-15 02:58:38
山西农业科学(2017年3期)2017-04-14 07:37:54
雷达学报(2017年6期)2017-03-26 07:53:04
天津医科大学学报(2015年2期)2015-12-22 09:24:32
食品工业科技(2014年13期)2014-03-11 18:16:51
