
环氧基POSS对PLA/PBAT共混材料发泡行为的影响
2017-12-06 02:57何仕成周洪福
中国塑料 2017年11期
刘 伟,何仕成,张 纯,任 粒,周洪福
(1.贵州理工学院材料与冶金工程学院,贵州 贵阳 550003;2.北京工商大学中国轻工业绿色塑料成型技术与质量评价重点实验室,北京 100048)
环氧基POSS对PLA/PBAT共混材料发泡行为的影响
刘 伟1,何仕成1,张 纯1,任 粒1,周洪福2
(1.贵州理工学院材料与冶金工程学院,贵州 贵阳550003;2.北京工商大学中国轻工业绿色塑料成型技术与质量评价重点实验室,北京100048)
通过熔融共混法制备了聚乳酸/聚己二酸对苯二甲酸丁二酯/聚倍半硅氧烷(PLA/PBAT/POSS)复合材料,并利用超临界二氧化碳(CO2)固相发泡法对复合材料进行发泡,通过差示扫描量热仪、高级动态流变仪和扫描电子显微镜等对复合材料的结晶行为、流变行为和发泡行为进行了研究。结果表明,POSS粒子对基体树脂具有增塑效应,PLA的冷结晶温度降低,结晶度提高;复合体系的流变性能明显提高,其发泡材料的泡孔密度和发泡倍率均随着POSS粒子含量的增加而增大,当加入7份(质量份,下同)POSS时,发泡材料的泡孔密度提高至8.25×107个/cm3,发泡倍率达到13倍;POSS粒子对PLA泡沫的泡孔形态具有显著的调控作用。
聚乳酸;聚己二酸对苯二甲酸丁二酯;笼形聚倍半硅氧烷;发泡;复合体系
0 前言
随着社会快速发展,研发高性能绿色高分子材料成为当前热点方向之一。其中,PLA与PBAT具有相似的加工性能和生物降解性,两者共混后可在较大范围内调控力学性能[1-3]。将PLA/PBAT两相高分子体系进一步发泡成型,得到的发泡材料具有应用于包装、生物医药和汽车等多个领域的巨大前景。然而,PLA和PBAT树脂可发性差、共混时两者相容性不好,所成型的发泡材料往往发泡倍率低、性能差,限制了其发展与应用[4-5]。为解决该问题,有研究采用了扩链法[6]、反应增容法[7]或无机粒子填充法[8]。在这些方法中,无机粒子填充法最为简单可行。通常,无机粒子表面不存在活性反应基团,只能在基体中简单分散,缺少对树脂的增容、扩链效果。因此,在无机粒子表面引入活性基团对解决低熔体强度问题有显著意义。多面体POSS是一类特殊的有机无机杂化结构填料,具有提高复合材料热稳定性、力学性能和阻燃性能等效果。此外,具有多环氧基团的POSS粒子与PLA和PBAT端基反应将得到支化结构产物,且反应产物对PLA/PBAT两相高分子体系的热性能和流变性能将产生影响,而这两方面影响作用是控制制备发泡材料的关键因素。
目前,PLA/PBAT/POSS复合材料的相关研究较少,特别是有关POSS粒子对PLA/PBAT的发泡行为及泡孔微观形态调控的研究。因此,本文采用熔融共混法制备了PLA/PBAT/POSS复合材料,研究了POSS粒子填充对PLA/PBAT两相高分子体系动态剪切流变性能、结晶性能和发泡行为的影响;研究工作的开展对于确定合理的工艺条件、探索POSS粒子在高分子发泡材料中的应用,以及制备高性能发泡材料具有重要意义。
1 实验部分
1.1 主要原料
PLA,4032D,美国Nature Works公司;
PBAT,Ecoflex C1200,德国BASF公司;
POSS,EP0409,环氧当量值为167,美国Hybrid Plastics公司。
1.2 主要设备及仪器
密炼机,XSS-60,上海科创橡塑机械设备有限公司;
超临界CO2固相发泡设备,自制;
精密鼓风干燥箱,BPG-9140A,上海一恒科学仪器有限公司;
差示扫描量热仪(DSC),Q100,美国TA仪器公司;
高级动态流变仪,MARS,美国赛默飞世尔科技有限公司;
扫描电子显微镜(SEM),Nova Nano 450,美国FEI公司;
样品表面喷金仪器,EMITECH K550X,捷克泰思肯仪器公司;
真密度测量仪,ULTRA-PYC,美国康塔仪器公司;
力学万能试验机,CMT6104,新三思计量技术有限公司;
数显冲击试验机,XJZ-50,承德试验机有限责任公司。
1.3 样品制备
PLA/PBAT/POSS复合材料的制备:将PLA和PBAT置于60 ℃干燥箱中干燥12 h去除原料表面水分;利用密炼机对PLA、PBAT和POSS粒子进行熔融共混,熔融共混温度为190 ℃,密炼时间为8 min,转速为60 r/min,共混配比如表1所示;熔融共混后的样品进一步干燥处理(干燥条件同上),以供流变性能、结晶性能测试和制备发泡样品之用;
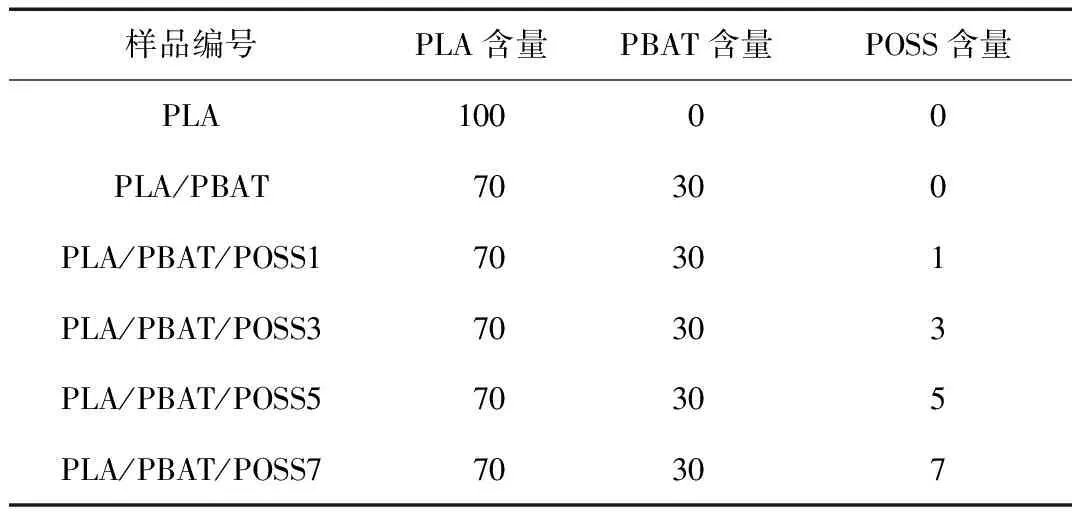
表1 PLA/PBAT/POSS复合材料的实验配方 份
PLA/PBAT/POSS发泡材料的制备:将PLA/PBAT/POSS复合材料置于高压釜内,升温至150 ℃,将CO2注入到高压釜内,并维持至20 MPa,使PLA/PBAT/POSS复合材料浸泡在超临界CO2中4 h,CO2分子逐渐溶解于复合材料中并达到平衡;随后,降温至130 ℃并释放釜内压力至常压,得到PLA/PBAT/POSS复合发泡材料,截取发泡芯层作测试样品。
1.4 性能测试与结构表征
结晶性能表征:利用DSC对PLA/PBAT/POSS复合材料的结晶和熔融行为进行表征;测试时样品处于氮气氛围中,以10 ℃/min的升温速率加热至190 ℃,观察其冷结晶和熔融行为;PLA/PBAT/POSS复合材料的结晶度(Xc)按式(1)计算:
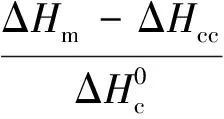
(1)
式中Xc——PLA的绝对结晶度,%
ΔHm——样品熔融焓,J/g
ΔHcc——样品的冷结晶焓,J/g
流变性能表征:对PLA/PBAT/POSS复合材料的动态剪切流变数据进行表征;试验样品置于直径为20 mm的圆形平行板间,测试间距为1 mm,测试温度为190 ℃,频率(ω)范围为0.1~100 s-1;其中,松弛时间谱通过式(2)进行计算:
(2)
式中H(t)——松弛时间谱,N/m2
G′——样品的储能模量,Pa
ω——动态剪切频率,rad/s
拉伸性能按GB/T 1040.1—2006进行测试,拉伸速率为5 mm/min,每种配方至少测试5根样条,并取平均值;
冲击强度按GB/T 1843—2008进行测试,测试使用带V形缺口的简支梁冲击方式,冲击能为2 J,每种配方至少测试5根样条,并取平均值;
泡孔形态表征:PLA/PBAT/POSS复合发泡材料的密度采用真密度测定仪进行测试,每组样品测试3次,取平均值作为该样品的密度;采用SEM对PLA/PBAT/POSS复合体系发泡材料的泡孔结构进行表征,将发泡样品浸泡在液氮中冷却并脆断,断面表面喷金,在放大倍率为500倍和5000倍下观察泡体的结构并计算泡孔密度(NC);NC通过计算机软件Image Tool进行分析,并通过式(3)进行计算:
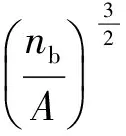
(3)
式中NC——泡孔密度,个/cm3
nb——统计面积中的泡孔数量,个
A——SEM照片中所选择的统计面积,cm2
ρ,ρf——未发泡样品和发泡后样品的密度,g/cm3
2 结果与讨论
2.1 结晶性能分析
从图1可以显著看出,随着POSS粒子含量的增加,PLA的冷结晶峰温度(Tcc)向低温方向移动。当POSS粒子加入量达到7份时,复合材料的Tcc由104.34 ℃降低至98.61 ℃。这种现象受控于POSS粒子对PLA的增塑效应,这种增塑效应从PLA的玻璃化转变温度降低也能够印证。由于增塑作用对分子链运动的影响,复合材料的结晶度也从0.7 %逐渐提高至2.4 %。Ash等[10]认为增塑作用的产生与填料和高分子的相互作用有关,且增塑效果与填料粒子的分散和含量有关,填料粒子比表面积越大,增塑现象越明显。结晶性能的变化对PLA/PBAT/POSS复合材料的可发性存在影响。在固相发泡过程中,CO2仅在无定形区域内溶解和扩散,而可发性是受控于结晶度的函数。此外,结晶区域有利于提高泡孔成核率,且在泡孔生长中阻碍发泡剂气体的逃逸,这种影响将最终决定发泡材料的泡孔尺寸、发泡倍率和泡孔形态[11]。此外,从图中还可看出,复合材料表现出熔融双峰特性,这种现象与复合材料的冷结晶过程有关[12]。POSS粒子的含量对复合材料熔融峰温度(Tm)的影响较小,熔融双峰温度维持在154 ℃和145 ℃左右。
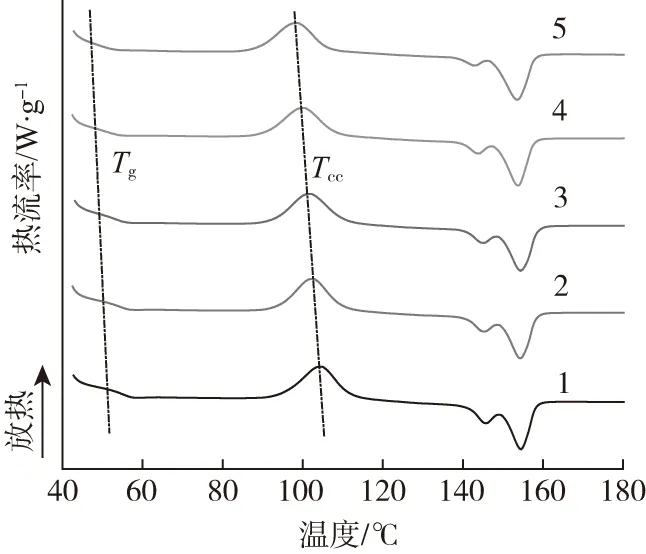
POSS含量/份:1—0 2—1 3—3 4—5 5—7图1 PLA/PBAT/POSS复合材料的DSC曲线Fig.1 DSC curves of the PLA/PBAT/POSS composites
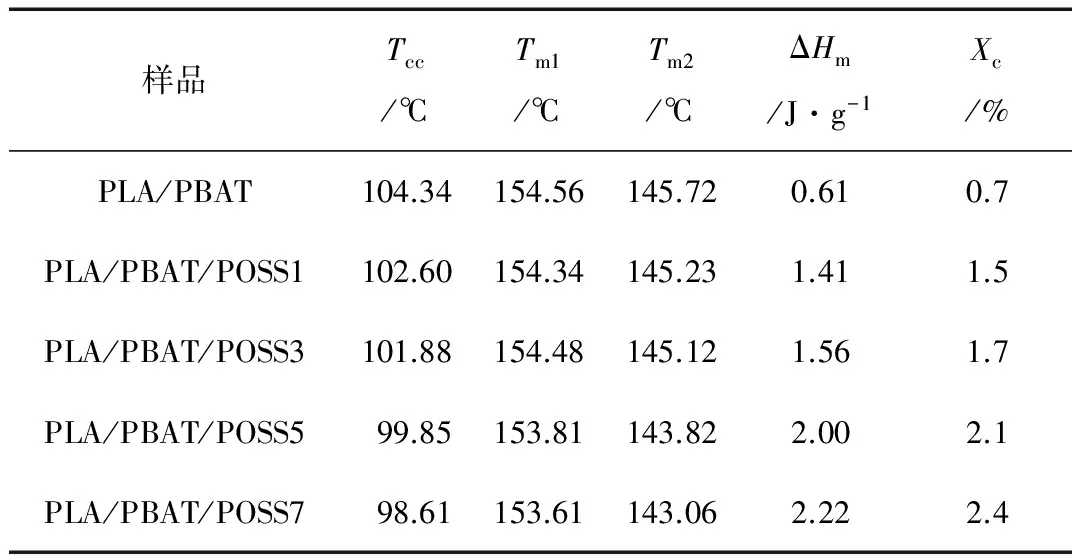
样品Tcc/℃Tm1/℃Tm2/℃ΔHm/J·g-1Xc/%PLA/PBAT104.34154.56145.720.610.7PLA/PBAT/POSS1102.60154.34145.231.411.5PLA/PBAT/POSS3101.88154.48145.121.561.7PLA/PBAT/POSS599.85153.81143.822.002.1PLA/PBAT/POSS798.61153.61143.062.222.4
2.2 流变性能分析
高分子材料的流变性能与其发泡行为密切相关,流变参数能够定量研究复合材料中POSS粒子含量与发泡行为之间的关系。但是,在实际发泡过程中,瞬时拉伸流变数据往往难以获取,通常采用间接流变手段,如动态剪切流变参数来表征发泡行为[13]。G′是复合材料黏弹性中弹性响应贡献部分,G′值越大,复合材料的熔体弹性贡献越大。从图2(a)中可以看出,所有复合材料样品的G′具有较强的ω依赖性。当只加入PBAT时,复合材料的G′值较纯PLA显著提高,这与PBAT本征熔体弹性较高有关。此外,在PLA/PBAT两相高分子体系的基础上,进一步加入POSS粒子,复合材料的G′值随着POSS粒子含量的增加而逐渐提高,这种现象说明了POSS粒子的环氧基团与分子链端基进行了反应,产生了具有一定支化结构的共聚物。在低频率区域,如果复合材料中具有支化结构,其动态黏弹参数通常会偏离经典的线性黏弹理论,即G′与ω的平方成正比,出现“第二平台”现象,这种现象是典型的末端区效应,说明加入POSS粒子有利于提高复合材料的熔体弹性。
从图2(b)中可以看出,复合材料的复数黏度(η*)随着ω的增加而降低,表现出典型的剪切变稀特征。在PLA/PBAT/POSS复合材料中,随着POSS粒子含量增加,复合材料的η*逐渐提高,这表明熔体流动性变得困难,产生这种现象是由于POSS粒子与分子链反应形成支化结构,限制了分子链热运动及分子链间相互穿梭置换,从而导致η*值提高。η*是发泡时泡孔结构形成的关键影响因素,当复合材料的η*值偏低时,泡孔易于发生“并泡”现象。因此,适当提高η*值将改善泡体结构。
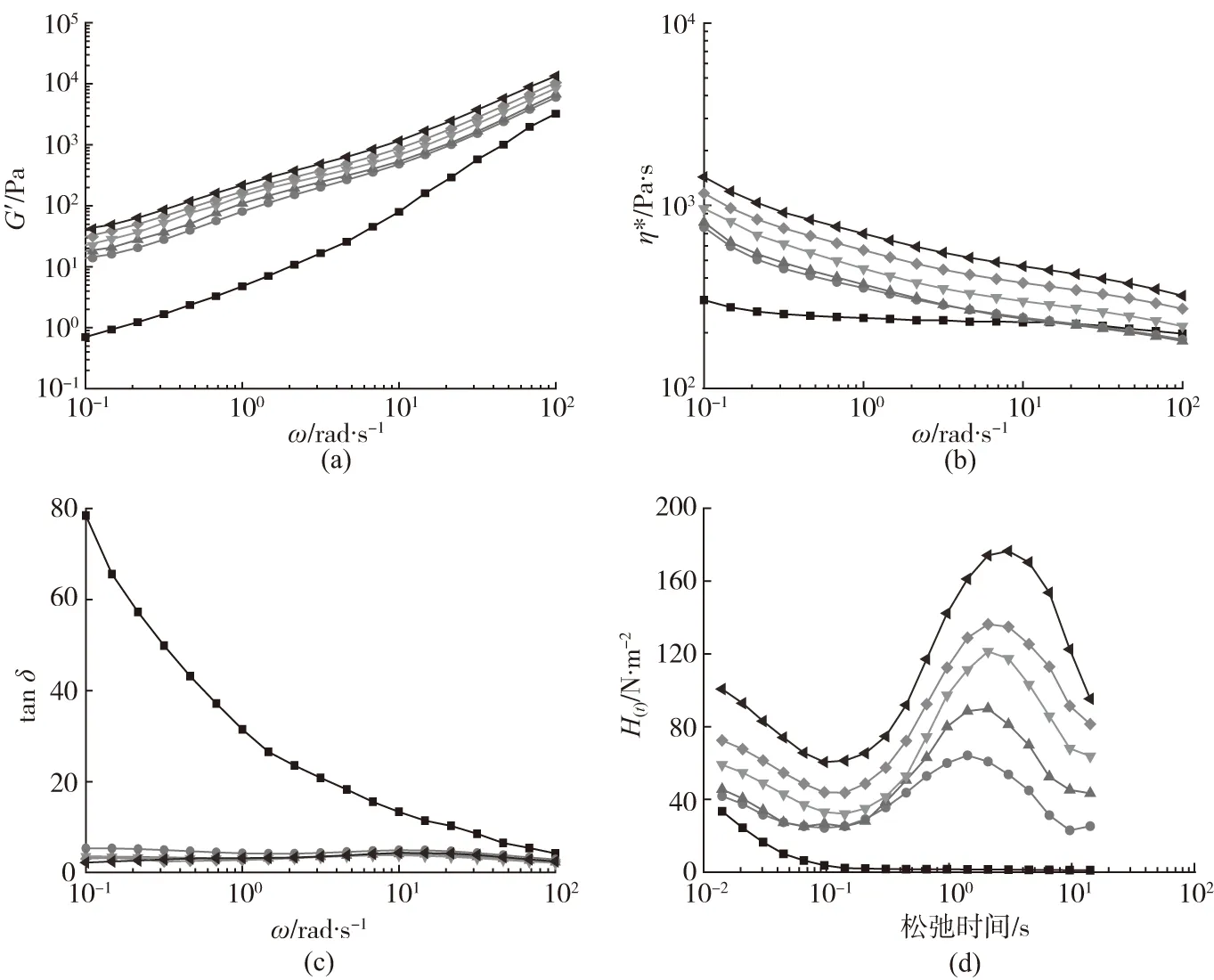
样品:■—PLA ●—PLA/PBAT ▲—PLA/PBAT/POSS1 ▼—PLA/PBAT/POSS3 ◆—PLA/PBAT/POSS5 ◀—PLA/PBAT/POSS7(a)G′-ω曲线 (b)η*-ω曲线 (c)tan δ-ω曲线 (d)松弛时间谱图图2 PLA/PBAT/POSS复合材料的流变曲线Fig.2 Rheological curves of PLA/PBAT/POSS composites
损耗因子(tanδ)是熔体在交变应力下,应变和所受到的应力相位差损耗角的正切值。tanδ越小,熔体弹性响应越快,高分子体系的可发性越好。从图2(c)中可以看出,加入PBAT或随POSS粒子含量的增加,复合材料末端区的tanδ值都显著降低。一般而言,tanδ值对复合材料的分子链结构变化十分敏感,同时也依赖于填料粒子的形态、尺寸、含量以及界面相互作用等因素。在PLA/PBAT/POSS复合材料体系中,POSS粒子与分子链发生反应,形成了一定支化程度的分子链结构。在末端区,这种支化结构受交变应力作用比较稳定,不易发生分子链解缠结,因此弹性响应明显,有利于维持泡孔结构稳定。
在研究高分子复合体系的黏弹性时,尤其是实际发泡过程中,需要表征以时间为变量的流变H(t)。从图2(d)中可以看出,所有样品在初始松弛时间阶段,随时间的延长,H(t)强度都逐渐降低,这是因为此时高分子链均由键长键角运动转变为链段运动。然而,随着POSS粒子含量的增加,复合材料的H(t)强度提高,且对应的松弛时间也小幅增加。在末端的松弛时间区域,加入POSS粒子的复合材料出现明显的“第二峰区”,且峰区强度受控于POSS粒子含量。这种现象的产生与分子链松弛过程变化有关,加入POSS粒子的复合材料不仅包括分子链之间的缠结松弛过程,同时还包括反应产生支化结构松弛过程,因此整体松弛时间更长;且此时的分子链运动过程相比更加复杂,导致H(t)强度上升。在发泡过程中,泡孔生长时泡孔壁经历剧烈的连续应变,而较长的松弛时间有利于维持材料结构的稳定,防止产生泡孔壁破裂和泡孔的合并现象。
2.3 力学性能分析
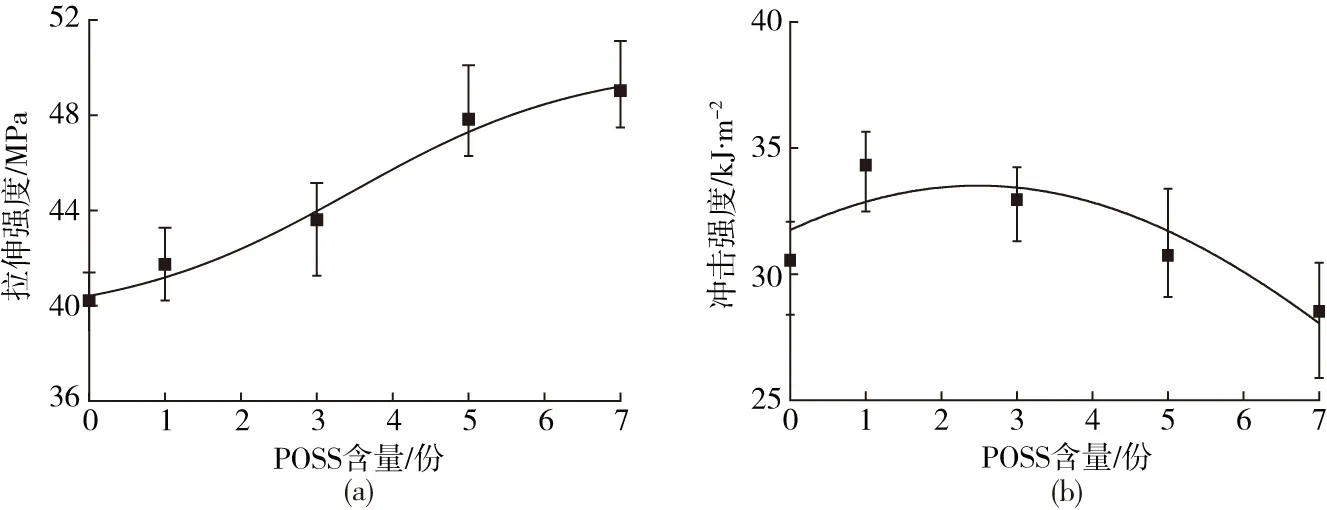
(a)拉伸性能 (b)冲击性能图3 PLA/PBAT/POSS复合材料的力学性能Fig.3 The relationship of mechanical properties of the PLA/PBAT composites and POSS content

(a)PLA/PBAT,×500 (b)PLA/PBAT/POSS1,×500 (c)PLA/PBAT/POSS3,×500 (d)PLA/PBAT/POSS5,×500(e)PLA/PBAT/POSS7,×500 (f)PLA/PBAT/POSS7,×5000图4 PLA/PBAT/POSS复合材料发泡样品的SEM照片Fig.4 SEM of PBAT/PBAT/POSS composite foams
从图3(a)中可以看出,PLA/PBAT复合材料的拉伸强度随POSS含量的增加呈“S形”增长,并在含量为1~5份时拉伸强度提高最明显,达到47 MPa,当继续提高POSS含量时,拉伸强度增加幅度变小。POSS粒子对复合体系的增强效应主要归因于其骨架效应和诱导结晶效应[14]。当POSS纳米粒子含量过低或过高时均无法起到较好的增强作用,这是因为含量较低时,粒子在复合材料中散落分布,对应力的消散作用和结晶诱导效果有限;而当含量较高时,其本身易产生团聚,较大的团聚颗粒容易引起应力集中,导致增强作用不明显。从图3(b)中可以看出,POSS粒子在低含量时对PLA/PBAT基体具有一定的增韧作用,当POSS含量为1份时,复合材料的冲击强度达到34 kJ/m2。根据粒子增韧机理,分散在PLA/PBAT基体中的POSS粒子能够阻碍冲击时裂纹的扩展,在裂纹前端面产生一定程度的弯曲,使冲击能量转换为弹性储能,从而起到增韧的效果[15]。但增韧效果也与粒子的尺寸、含量和几何形状有关。PLA/PBAT复合材料的增韧受控于POSS粒子的分散效果,高含量产生的团聚不利于增韧。此外,随着POSS粒子含量增加,体系的结晶度提高,断裂模式由韧性断裂向脆性断裂变化[16]。因此,随着POSS粒子含量进一步提高,复合材料的增韧效果降低。
2.4 发泡性能分析
从图4中可以看出,PLA/PBAT两相共混体系的泡孔结构并不均匀,泡孔呈椭圆形,具有一定程度的泡孔合并现象。这主要是因为两相高分子的简单共混未改变分子链结构,同时,PLA和PBAT树脂本身可发性较差,从而共混体系的整体可发性并未改善,而这种现象在动态流变行为研究中也得到了印证。然而,在PLA/PBAT共混体系中加入POSS粒子后,泡孔合并和坍塌的现象减少,当POSS粒子含量超过3份时,泡孔形态显著改善,且发泡后的POSS粒子分散在泡孔壁上。泡孔形态的改善趋势与复合体系的松弛过程转变有关。根据前述研究结果,填充POSS粒子有效地提高了末端区的流变性能,在POSS粒子周围的分子链形成支化结构,改变了复合体系的H(t),延长了高分子的弛豫过程,体系的黏弹性得到提高,从而可发性提高。
由表3可知,由于PLA和PBAT树脂的黏弹性不适于发泡成型, 其共混材料发泡后泡孔坍塌、合并,导致发泡倍率和NC偏低。随着POSS粒子的引入,复合材料的流变性能得到改善,其发泡倍率由3.71倍提高至13倍。此外,泡孔数量也随着POSS粒子含量增加而显著提高,NC由1.27×107个/cm3提高至8.25×107个/cm3,泡孔尺寸也由84.63 μm降低至30.73 μm,这与POSS粒子的异相成核作用促进泡孔大量成核有关。

表3 PLA/PBAT/POSS复合发泡材料的发泡性能Tab.3 Foaming properties of PLA/PBAT/POSScomposite foams
3 结论
(1)POSS粒子具有增塑效应,降低了复合材料的Tcc,同时提高了其Xc;POSS粒子具有增强作用,当POSS含量达到5份时,复合材料的拉伸强度达到47 MPa左右;另一方面,POSS粒子含量为1份时,对复合材料具有一定增韧效果,冲击强度达到34 kJ/m2,但进一步提高POSS含量时,复合材料的冲击强度降低;
(2)POSS粒子能够与分子链反应形成具有支化的分子链结构,有效提高了复合材料的熔体弹性、η*,并降低了tanδ;
(3)POSS粒子有利于提高复合材料的发泡倍率,优化泡孔结构,发泡后POSS粒子均匀分散在泡孔壁上,因此POSS粒子对PLA发泡过程和泡孔结构具有显著的调控作用。
[1] Nofar M, Maani A, Sojoudi H, et al. Interfacial and Rheological Properties of PLA/PBAT and PLA/PBSA Blends and Their Morphological Stability Under Shear Flow[J]. Journal of Rheology, 2015, 59(2):317-333.
[2] Nofar M, Heuzey M C, Carreau P J, et al. Effects of Nanoclay and Its Localization on the Morphology Stabilization of PLA/PBAT Blends Under Shear Flow[J]. Polymer, 2016, 98:353-364.
[3] Arruda L C, Magaton M, Bretas R E S, et al. Influence of Chain Extender on Mechanical, Thermal and Morphological Properties of Blown Films of PLA/PBAT Blends[J]. Polymer Testing, 2015, 43:27-37.
[4] Kuo C C, Liu L C, Liang W C, et al. Preparation of Polylactic Acid(PLA) Foams with Supercritical Carbon Dioxide and Their Applications for Reflectors of White Light-emitting Diode(LED) Lamps[J]. Materials Research Bulletin, 2015, 67:170-175.
[5] Kim S, Park S W, Lee K. Fishing Performance of Environmentally Friendly Tubular Pots Made of Biodegradable Resin(PBS/PBAT) for Catching the Conger Eel Conger Myriaster[J]. Fisheries Science, 2014, 80(5):887-895.
[6] Schneider J, Manjure S, Narayan R. Reactive Modification and Compatibilization of Poly(lactide) and Poly(buty-lene adipate-co-terephthalate) Blends with Epoxy Functio-nalized-poly(lactide) for Blown Film Applications[J]. Journal of Applied Polymer Science, 2016, 133(16):1-9.
[7] Kumar M, Mohanty S, Nayak S K, et al. Effect of Glycidyl Methacrylate(GMA) on the Thermal, Mechanical and Morphological Property of Biodegradable PLA/PBAT Blend and Its Nanocomposites[J]. Bioresource Technology, 2010, 101(21):8406-8415.
[8] Dil E J, Favis B D. Localization of Micro- and Nano-silica Particles in Heterophase Poly(lactic acid)/Poly(butylene adipate-co-terephthalate) Blends[J]. Polymer, 2015, 76:295-306.
[9] Herrera Ricard, Franco Lourdes, Puiggali Jordi. Characterization and Degradation Behavior of Poly(butylene adipate-co-terephthalate)s[J]. Journal of Polymer Science Part A:Polymer Chemistry, 2002, 40(23):4141-4157.
[10] Ash B J, Siegel R W, Schadler L S.B J Ash, R W Siegel and L S Schadler, ‘Glass-transition Temperature Beha-vior of Alumina/PMMA Nanocomposites’[J]. Journal of Polymer Science Part B: Polymer Physics, 2005, 43(1):114-114.
[11] Ding Weidan, Kuboki T, Wong Anson, et al. Rheology, Thermal Properties, and Foaming Behavior of High D-content Polylactic Acid/Cellulose Nanofiber Compo-sites[J]. Rsc Advances, 2015, 5(111):91544-91557.
[12] Santis F D, Pantani R, Titomanlio G. Nucleation and Crystallization Kinetics of Poly(lactic acid)[J]. Thermochimica Acta, 2011, 522(1/2):128-134.
[13] Najafi N, Heuzey M C, Carreau P J, et al. Rheological and Foaming Behavior of Linear and Branched Polylactides[J]. Rheologica Acta, 2014, 53(10):779-790.
[14] Liang Jizhao. Reinforcement and Quantitative Description of Inorganic Particulate-filled Polymer Composites[J]. Composites Part B: Engineering, 2013, 51(4):224-232.
[15] Jazi S H S, Bagheri R, Esfahany M N. The Effect of Surface Modification of(Micro/Nano)-calcium Carbonate Particles at Various Ratios on Mechanical Properties of Poly(vinyl chloride) Composites[J]. Journal of Thermoplastic Composite Materials, 2015, 28(4): 479-495.
[16] 董金虎. 聚合物基复合材料的增强增韧[J]. 中国塑料, 2012,26(7):20-27.
Dong Jinhu. Toughening and Strengthening of Polymeric Composites[J]. China Plastics, 2012,26(7):20-27.
EffectofEpoxy-basedPOSSonFoamingBehaviorofPLA/PBATBlends
LIUWei1,HEShicheng1,ZHANGChun1,RENLi1,ZHOUHongfu2
(1.School of Materials and Metallurgical Engineering, Guizhou Institute of Technology, Guiyang550003, China;2.Key Laboratory of Processing and Quality Evaluation Technology of Green Plastics of China NationalLight Industry Council, Beijing Technology and Business University, Beijing100048, China)
Poly(lactic acid) (PLA)/poly(butyleneadipate-co-terephthalate) (PBAT)/polyhedral oligomeric silsesquioxane (POSS) composites and their foams were prepared by a melt-mixing method and a supercritical CO2solid state foaming method, respectively. Thermal behavior, rheological property, and foaming behavior of the composites were studied. The results confirmed the plasticization effect of POSS particles on the composites. The rheological results indicated that melt elasticity of the composites was improved with the addition of POSS. As a result, cellular structure of the composites foams was enhanced. Cell density and foaming expansion ratio of the composite foams increased with an increase of POSS content. The composite foams achieved a cell density of 8.25×107cell/cm3and an expansion ratio of 13 at the POSS content of 7 phr. POSS was found to have a positive effort on controlling the cellular structure of the composite foams.
poly(lactic acid); poly(butyleneadipate-co-terephthalate); polyhedral oligomeric silses-quioxane; foam; composite
2017-06-19
国家自然科学基金(51703039);贵州省社发攻关项目(2001004036014);贵州省科技厅联合基金(黔科合LH字[2014]7372号)
联系人,lichpistol@126.com
TQ328.9
B
1001-9278(2017)11-0053-07
10.19491/j.issn.1001-9278.2017.11.008
猜你喜欢
功能高分子学报(2022年5期)2022-10-19
沈阳工业大学学报(2022年5期)2022-10-06
中国塑料(2022年5期)2022-06-09
工程塑料应用(2022年4期)2022-04-23
包装工程(2022年1期)2022-01-26
科教导刊·电子版(2021年6期)2021-05-06
潍坊学院学报(2020年6期)2020-11-22
航空制造技术(2020年18期)2020-10-31
陶瓷学报(2019年6期)2019-10-27
表面工程与再制造(2019年3期)2019-09-18
