
丙型肝炎病毒感染过程中其NS3/4A蛋白酶切割线粒体抗病毒信号蛋白的动态过程
2017-11-01 06:10许军杜晓婷张扬袁正宏易志刚
微生物与感染 2017年5期
许军,杜晓婷,张扬,袁正宏,易志刚
复旦大学基础医学院病原生物学系,医学分子病毒学教育部/卫生部重点实验室,上海 200032
·论著·
丙型肝炎病毒感染过程中其NS3/4A蛋白酶切割线粒体抗病毒信号蛋白的动态过程
许军,杜晓婷,张扬,袁正宏,易志刚
复旦大学基础医学院病原生物学系,医学分子病毒学教育部/卫生部重点实验室,上海 200032
已知丙型肝炎病毒(hepatitis C virus,HCV)可通过其蛋白酶NS3/4A切割线粒体抗病毒信号蛋白(mitochondrial antiviral signaling protein,MAVS)来逃逸天然免疫识别,但尚不清楚其切割动力学及切割在抑制干扰素中的作用。本研究旨在细胞模型中探讨HCV感染过程中病毒复制建立及病毒NS3/4A切割MAVS的动态过程,探究NS3/4A切割MAVS对病毒逃逸宿主天然免疫建立感染的贡献。首先构建基于绿色荧光蛋白(green fluorescent protein,GFP)的MAVS切割报告系统(GFP-NLS-MAVS-TM462),用 HCV Jc1-Gluc 感染Huh7.5/GFP-NLS-MAVS-TM462细胞。结果显示,病毒复制早期MAVS切割效率较低;NS3/4A高效切割MAVS发生于HCV复制晚期,且其切割效率与NS3蛋白水平相关。利用带有GFP ypet的HCV报告病毒Jc1-378-1感染Huh7.5/RFP-NLS-MAVS-TM462细胞,在单细胞水平观察HCV感染阳性细胞中MAVS被切割情况,发现HCV复制细胞中仅部分细胞MAVS被切割。进一步研究发现,不同基因型NS3/4A切割MAVS的效率仅与NS3表达水平相关。以上结果提示,HCV蛋白酶NS3/4A切割MAVS依赖NS3/4A蛋白在病毒复制过程中的累积,对在病毒复制早期逃逸宿主天然免疫建立感染可能无显著贡献。
丙型肝炎病毒;非结构蛋白3/4A;线粒体抗病毒信号蛋白;干扰素
丙型肝炎病毒(hepatitis C virus,HCV)是单股正链RNA病毒,隶属于黄病毒科丙型肝炎病毒属[1]。HCV感染人体后导致70%~80%感染者慢性化,其中部分慢性化患者发展为肝硬化、肝细胞肝癌[2-4]。HCV基因组编码一多肽蛋白前体,其经宿主细胞信号肽酶和病毒蛋白酶加工成结构蛋白和非结构蛋白(nonstructural protein,NS)[5-6]。其中NS3为双功能酶,具有丝氨酸蛋白酶活性和解旋酶活性[7],发挥丝氨酸蛋白酶活性需NS4A作为辅因子。
一般认为,病毒感染后机体天然免疫系统通过位于细胞膜或细胞内的模式识别受体(pattern recognition receptor,PRR)识别病毒特定的病原相关模式分子(pathogen-associated molecular pattern,PAMP),启动抗病毒细胞因子如干扰素等的合成。PRR包括视黄酸诱导基因Ⅰ样受体(retinoic acid inducible gene Ⅰ like receptor,RLR)和Toll样受体(Toll-like receptor,TLR)等[8],其中视黄酸诱导基因Ⅰ(retinoic acid inducible gene Ⅰ,RIG-Ⅰ)和黑色素瘤分化相关蛋白5(melanoma differentiation-associated protein 5, MDA5)均属RLR。RLR识别PAMP后,招募其接头分子线粒体抗病毒信号蛋白(mitochondrial antiviral signaling protein,MAVS)激活下游信号分子,诱导抗病毒蛋白和各种细胞因子如Ⅰ型、Ⅲ干扰素等表达,从而发挥抗病毒作用[2,9]。早期研究报道,HCV可通过其丝氨酸蛋白酶NS3/4A切割MAVS第508位半胱氨酸(cysteine,Cys),使MAVS从线粒体膜上脱落,从而阻断干扰素信号通路,提示HCV可能通过切割MAVS逃逸干扰素作用,导致病毒持续性感染[10]。但近期研究报道,HCV感染原代肝细胞[11]和表达有CD81/miR122的 HepG2细胞均可诱生干扰素[12];且过表达MAVS或MAVS-C508R突变体(对NS3/4A蛋白酶有抗性)均可抑制HCV复制,两者抑制HCV复制的能力没有显著差异[13],提示NS3/4A介导的MAVS切割在HCV感染细胞中能否阻断干扰素产生值得探讨。本研究通过构建MAVS切割报告系统,发现NS3/4A高效切割MAVS发生于病毒复制晚期。进一步研究发现,不同基因型NS3/4A切割MAVS的切割效率无显著差异,仅与病毒NS3表达水平相关。以上结果提示,HCV 蛋白酶NS3/4A切割MAVS依赖NS3/4A蛋白在病毒复制过程中的累积,对病毒复制早期逃逸宿主天然免疫建立感染可能无显著贡献。
1 材料与方法
1.1 材料
肝癌细胞系Huh7、HCV亚基因组sgJFH1复制子细胞[14]和BB7复制子细胞[15]为本室保存。肝癌细胞系Huh7.5、Jc1G HCV细胞培养体系(HCV cell culture,HCVcc)[16]及带有ypet绿色荧光蛋白(green fluorescent protein,GFP)报告基因的单顺反子HCV感染性克隆Jc1-378-1均由美国洛克菲勒大学HCV研究中心Charles Rice 教授赠予[14]。人胚肾细胞HEK293T购自中国科学院上海生命科学研究院。胎牛血清购自Biological Industries,DMEM培养基购自Gibco,Renilla luciferase substrate购自Promega(E2820)。pV1-BSD-GFP-NLS-MAVS-TM462 和pTRIP-RFP-NLS-MAVS-TM462等质粒为本课题组构建[17]。实验中所用抗体及来源如下:NS5A抗体(9E10),NS3抗体(Virogen;217-A),辣根过氧化物酶(horseradish peroxidase,HRP)标记羊抗鼠IgG (Santa Cruz;sc-2005),抗β-actin抗体 (Sigma;A1978),GFP抗体(Santa Cruz;sc-9996),MAVS抗体 (Santa Cruz;sc-166583)。
1.2 方法
1.2.1细胞培养肝癌细胞系Huh7、 Huh7.5,HCV亚基因组复制子细胞sgJFH1和BB7,人胚肾细胞HEK293T等均用含10%胎牛血清的DMEM培养基,于37 ℃、5% CO2恒温恒湿培养箱中培养,每2~3 d进行细胞传代。Huh7.5细胞培养基中加入0.01 mmol/L非必需氨基酸。HCV亚基因组复制子细胞sgJFH1和BB7培养基中加入0.5 μg/mL杀稻瘟菌素(blasticidin)。
1.2.2十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodiumdodecylsulfate-polyacrylamidegelelectrophoresis,SDS-PAGE)和蛋白免疫印迹法细胞样品用含SDS的样品缓冲液处理,沸水浴10 min。蛋白电泳和转膜采用Bio-Rad装置进行。配制4.5%浓缩胶和10%凝胶,恒流15 mA进行电泳。电泳后,将蛋白转移到硝酸纤维素膜上,与相应蛋白的抗体4 ℃孵育过夜,再与HRP标记的二抗室温孵育2 h,用增强化学发光法(enhanced chemiluminescence,ECL)检测。
1.2.3DNA转染将Huh7.5细胞以2.0×105/mL铺于24孔板中,16~18 h后进行转染。取50 μL opti-MEM,加入待转染质粒0.5 μg,振荡混匀,静置5 min;向其中加入1.5 μL X-tremeGENE HP DNA Transfection Reagent 脂质体,振荡混匀,静置15 min;将混合物均匀缓慢滴加到已更换新鲜细胞培养液的24孔板中,转染后8 h更换培养基。
1.2.4慢病毒介导的稳定细胞株的构建将含有目的基因的慢病毒质粒、VSVG、HIV-gagpol按4∶1∶4比例共转染HEK293T细胞进行慢病毒包装,8 h后更换新鲜培养基。转染后48 h,收集含有慢病毒的细胞上清液,用0.45 μm过滤器过滤,并加入8 μg/mL polybrene和20 mmol/L HEPES。将含有慢病毒的细胞上清液转导细胞,8 h后更换新鲜培养基,48 h后换成含5 μg/mL杀稻瘟菌素的培养基筛选1~2周,用含0.5 μg/mL杀稻瘟菌素的培养基维持培养。
1.2.5HCVcc的制备(以Jc1-Gluc为例) HCVcc由体外转录HCV RNA电转Huh7.5 细胞获得,具体步骤见文献[18]。简要介绍如下:用XbaⅠ限制性内切酶将质粒Jc1-Gluc线性化并回收,T7体外转录试剂盒进行RNA体外转录,QIAGEN RNA纯化试剂盒纯化RNA。将10 μg体外转录的Jc1-Gluc RNA电转至6×106个Huh7.5细胞,细胞贴壁后换成新鲜培养基,每隔2 d收集含有HCV感染性颗粒的细胞上清液,用0.45 μm过滤器过滤后分装冻存。
1.2.6HCVcc滴度测定(有限稀释法) 将Huh7.5细胞以8×104/mL接种于96孔板中。次日细胞贴壁后,将HCV进行10倍梯度稀释,用不同稀释度的病毒感染Huh7.5细胞。感染后12 h更换新鲜培养基,3~4 d后用免疫荧光法标记HCV NS5A并计数阳性细胞,根据 Reed-Muench方法计算半数组织培养感染剂量(50% tissue culture infective dose,TCID50)。具体方法见文献[19]。
1.3 统计学分析
采用Graphpad Prism 6软件作图并分析数据,数据以mean±SD表示。采用非配对t检验比较两组间差异,P<0.05为差异有统计学意义。
2 结果
2.1NS3/4A高效切割MAVS发生于病毒复制晚期
为研究HCV感染过程中NS3/4A切割MAVS的时效性,即HCV复制建立过程中病毒蛋白尤其是NS3蛋白的表达水平、病毒复制水平及MAVS切割情况之间的动态关系,构建MAVS切割报告系统(pV1-BSD-GFP-NLS-MAVS-TM462)(图1A)。该质粒含MAVS第462~540位氨基酸,包括MAVS跨膜区及NS3/4A蛋白酶的切割位点508-Cys,因此与全长MAVS定位一致,且能被NS3/4A蛋白酶切割[20]。GFP和核定位信号肽(nuclear localization signal,NLS)位于N端,MAVS被切割后,NLS将引导含有GFP的融合蛋白由胞质线粒体转入细胞核。
用Jc1-Gluc[21]感染Huh7.5/GFP-NLS-MAVS-TM462细胞(Jc1-Gluc为HCV感染性克隆,HCV基因组中插入可分泌的荧光素酶Gluc),在不同时间点检测Gluc活性(图1B)来反映病毒复制水平,并用蛋白免疫印迹法(图1C)和荧光显微镜(图1E)观察MAVS-TM462被切割情况。结果显示,HCV在感染后第3天病毒复制报告基因Gluc基本不再上升(图1B),提示病毒复制趋于稳定。利用蛋白免疫印迹法,发现NS3于病毒感染后第2天即能明显检测到,随后增加,第5天表达水平最高(图1C)。从病毒感染后第2天开始,能明显观察到MAVS切割报告蛋白GFP-TM462被切割的条带(Cl),随后增加(图1C)。前4 d内,GFP-TM462切割效率(被切割条带Cl/未被切割条带FL)较低,切割效率均<3.0%(图1D)。病毒感染后第5天,NS3表达增强,同时GFP-TM462切割效率升至16.5%(图1C、1D)。荧光显微镜下,病毒感染后第3天明显看到GFP入核(箭头所示),随后入核细胞增加(图1E)。上述结果表明,病毒感染状态下MAVS的切割效率与病毒蛋白酶NS3的表达相关,切割效率在病毒复制晚期随NS3的累积而增强。

A: Schematic representation of MAVS and GFP-NLS-MAVS-TM462. MAVS, a 540-amino acid protein, contains a caspase activation and recruitment domain (CARD), a proline-rich region, and a C-terminal transmembrane domain (TM). GFP-NLS-MAVS-TM462 contains N-terminal green fluorescent protein (GFP), nuclear localization signal peptide (NLS) and C-terminal 462-540 amino acids of MAVS, which contains TM of MAVS and NS3/4A cleavage site 508-Cys. B-E: Huh7.5/GFP-NLS-MAVS-TM462 cells were infected with Jc1-Gluc (MOI=0.5). Cells were passaged at 4 d post infection. At 24, 48, 72 and 96 h post infection, the supernatants and cells were harvested. B: Gaussia luciferase (Gluc) activity in the supernatants from infected cells. mean±SD,n=3. C: Cell lysates of Huh7.5/GFP-NLS-MAVS-TM462 infected with Jc1-Gluc were analyzed by Western blotting. Mock, uninfected Huh7.5 cells; Control, uninfected Huh7.5/GFP-NLS-MAVS-TM462 cells. “FL” indicates the full length of MAVS-TM462 and “Cl” indicates the cleaved bands of MAVS-TM462. D: The bands of “FL” and “Cl” were quantified and the cleavage efficiency was calculated. E: Huh7.5/GFP-NLS-MAVS-TM462 infected with Jc1-Gluc observed by fluorescence microscopy. The white arrows indicate the cells with GFP-TM462 cleaved and translocated to the nucleus.
图1NS3/4A高效切割MAVS发生于HCV复制晚期
Fig.1NS3/4A-mediatedMAVScleavageoccurredatthelatetimeofHCVreplication
2.2在HCV感染阳性细胞中仅部分细胞MAVS被完全切割
为在单细胞水平观察HCV感染与MAVS被切割情况,进一步利用Jc1-378-1感染Huh7.5/pTRIP-RFP-NLS-MAVS-TM462细胞株。Jc1-378-1的 NS5A区段同框融合ypet GFP报告基因,HCV复制时产生融合有ypet的NS5A[14];而pTRIP-RFP-NLS-MAVS-TM462质粒与 pV1-BSD-GFP-NLS-MAVS-TM462相似,可通过观察RFP是否入核反映MAVS是否被切割。荧光显微镜观察显示,病毒感染后第2天可检测到ypet阳性细胞及RFP入核细胞(白色箭头所示)(图2A)。随后ypet阳性细胞及RFP入核细胞逐步增加,表明随着HCV复制,MAVS开始发生切割(图2A);ypet阳性细胞中也能观察到RFP未入核细胞(红色箭头所示)(图2A)。统计结果显示,感染后不同时间点RFP阳性细胞中RFP入核细胞所占比例小于ypet阳性细胞比例(图2B、2C)。HCV感染后第2天,RFP阳性细胞中有8.8% HCV感染阳性(ypet阳性)细胞,其中仅1.5%的细胞中MAVS发生切割 (RFP入核);而HCV感染后第5天,HCV感染阳性细胞比例达13.1%,MAVS发生切割的细胞比例也只有6%,即约50%的HCV感染阳性细胞中MAVS发生切割(图2B、2C)。以上结果表明,HCV感染阳性细胞中,仅部分细胞MAVS被完全切割。
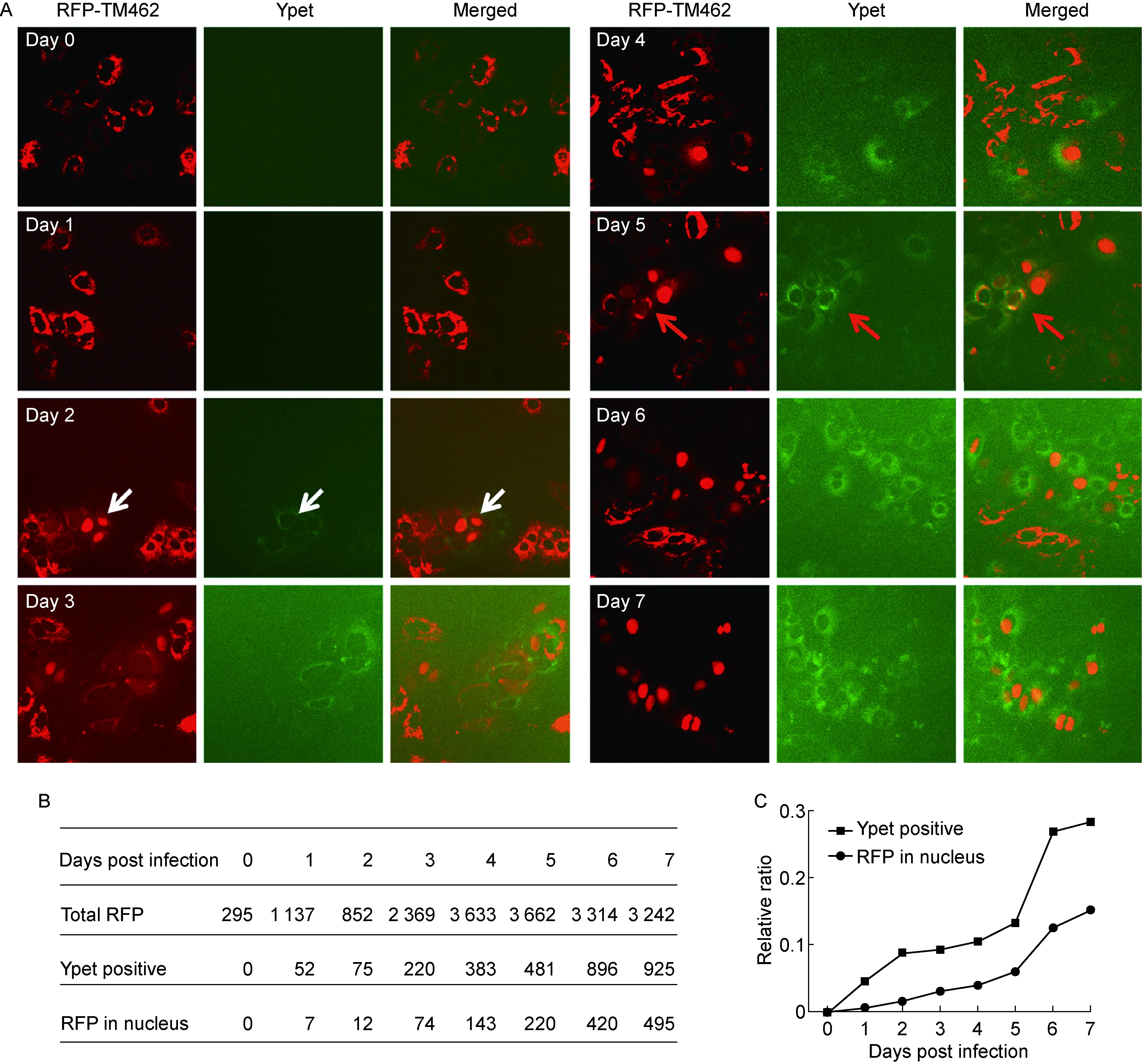
A: Huh7.5/RFP-NLS-MAVS-TM462 cells were infected with JC1-378-1 (MOI=0.1) and the cells were observed by fluorescence microscopy at various time points post infection. The white arrow indicates HCV-infected cells (ypet-positive) with RFP-TM462 cleaved and translocated to the nucleus (RFP in nucleus). The red arrow indicates HCV-infected cells with RFP-TM462 intact and remained in the cytoplasm. B: Total quantified cell numbers of the RFP positive cells (total RFP), HCV-infected cells (ypet-positive) and the cells with MAVS cleavage (RFP in the nucleus) at various time points post infection. C: The percentages of the HCV-infected cells (ypet-positive) and the cells with MAVS cleavage (RFP in nucleus) are plotted.
图2HCV复制阳性细胞中仅部分细胞MAVS被切割
Fig.2MAVSwascleavedonlyinpartoftheHCV-infectedcells
2.3不同基因型NS3/4A对MAVS的切割无显著差异
为探讨不同基因型NS3/4A切割MAVS是否有差异,将含有pV1-BSD-GFP-NLS-MAVS-TM462的慢病毒转导1b基因型复制子细胞BB7和2a基因型复制子细胞sgJFHI,通过蛋白免疫印迹法(图3A)和荧光显微镜(图3C)观察 MAVS被切割情况。结果显示,在1b型和2a型复制子细胞中,MAVS-TM462均能被有效切割,且大部分细胞中GFP发生入核(图3A、3C)。定量分析发现,2a型复制子细胞中MAVS-TM462被切割效率(78%)高于1b型复制子细胞中MAVS被切割效率(65%)(图3B)。必须注意,NS3表达量在2a型复制子细胞中亦高于1b型复制子细胞(图3A)。为克服可能由于病毒复制水平差异导致两种不同基因型蛋白酶NS3的表达水平差异,进一步在Huh7/GFP-NLS-MAVS-TM462细胞株中过表达不同基因型NS3/4A,观察MAVS切割情况。在过表达条件下,两种不同基因型NS3蛋白表达量基本一致,且MAVS-TM462均能被有效切割(图4A)。 定量分析显示,1b与2a型NS3/4A切割MAVS-TM462的效率(分别为56%和59%)无显著差异(图4B)。荧光显微镜观察显示,转染细胞中大部分GFP发生入核(图4C)。以上结果提示,不同基因型NS3/4A切割MAVS的切割效率可能仅与NS3蛋白的表达水平相关。

BB7 or sgJFH1 cells were transduced with lentivirus of GFP-NLS-MVAS-TM462. A: The cell lysates were analyzed by Western blotting with indicated antibodies. “FL” indicates the full length of MAVS-TM462 and “Cl” indicates the cleaved bands of MAVS-TM462. B: The MAVS cleavage efficiency in A was calculated (n=3,t-test,*P<0.05). C: The cells were observed by fluorescence microscopy.
图3 1b型和2a型复制子细胞中NS3/4A切割MAVS效率与NS3蛋白水平相关
Fig.3ThecleavageefficiencywascorrelatedwiththeproteinlevelofHCVNS3/4Ain1band2arepliconcells
3 讨论
HCV复制过程中,丝氨酸蛋白酶NS3/4A切割MAVS被认为是HCV逃逸干扰素作用的重要途径。亦有报道HCV感染原代肝细胞[11]及特定的经基因修饰的肝癌细胞系[12]均可诱导干扰素产生;在过表达 MAVS-C508R突变体与野生型MAVS状态下,两者抑制HCV复制的能力无显著差异,提示 NS3/4A切割MAVS在介导HCV逃逸干扰素过程中的作用还有待进一步研究。
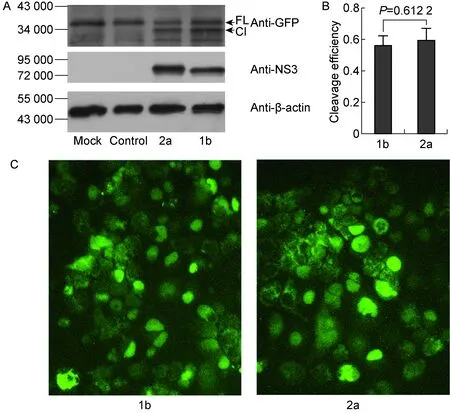
Huh7/GFP-NLS-MAVS-TM462 cells were transfected with pcDNA3.1-NS3/4A (genotypes 1b and 2a). At 48 h post transfection, the cells were harvested. A: The cell lysates were analyzed by Western blotting with the indicated antibodies. “FL” indicates the full length of MAVS-TM462 and “Cl” indicates the cleaved bands of MAVS-TM462. B: The MAVS cleavage efficiency in A was calculated (n=3,t-test). C: The cells were observed by fluorescence microscopy.
图4过表达不同基因型NS3/4A(1b型和2a型)对MAVS切割无显著差异
Fig.4TherewasnodifferenceinMAVScleavagebyover-expressionofNS3/4Afromgenotypes1band2a
本研究构建了基于GFP的MAVS切割报告系统,利用该报告系统发现NS3/4A高效切割MAVS发生于HCV复制晚期。HCV感染阳性细胞中,仅部分细胞的MAVS被切割。NS3/4A能以顺式切割(cis)及反式 (trans)切割方式加工病毒多肽[7],但一般认为顺式切割效率不依赖蛋白酶浓度,而反式切割效率依赖NS3蛋白酶浓度。与加工病毒蛋白不同,NS3/4A只能以反式切割方式切割MAVS,可能只有NS3/4A蛋白积累到一定量方能达到最大切割效率,因此本研究仅在病毒复制晚期观察到高效的MAVS切割(图1)。
那么,在感染早期HCV能否逃逸机体先天免疫识别?RIG-Ⅰ已被证明参与HCV的天然免疫识别[22-23],由于Huh7.5细胞中RIG-Ⅰ通路缺失,HCV可在其中高复制(high permissiveness)[18],提示RIG-Ⅰ介导的天然免疫对HCV复制的限制。近期有报道除RIG-Ⅰ外,MDA5 亦参与HCV的天然免疫识别[24-25]。Huh7.5.1细胞中过表达MAVS显著降低HCV复制水平[13],提示MAVS介导的信号通路也限制HCV复制[2,8-9]。综上所述,HCV可被RIG-Ⅰ或MDA5识别,且RIG-Ⅰ/MDA5-MAVS信号通路对其复制具有限制作用。鉴于本研究显示HCV NS3/4A蛋白酶切割MAVS主要发生于病毒复制晚期,可推测在病毒复制早期,需通过其他机制介导HCV逃逸宿主天然免疫。严重急性呼吸综合征(severe acute respiratory syndrome,SARS)病毒、西尼罗河病毒、脊髓灰质炎病毒、雀麦花叶病毒及HCV等正链RNA病毒的复制均发生于膜相关的复制复合体内。此类病毒复制复合体均位于宿主细胞的特定膜结构中,由病毒蛋白对宿主膜结构进行改造而形成一个有包膜的相对封闭的囊泡结构(membranous web)[26-30]。此囊泡结构可能隔离病毒复制产物,阻止病毒复制过程中产生的危险信号被天然免疫识别分子识别。免疫荧光结果显示,HCV RNA由于复制复合体的存在,使RIG-Ⅰ及MDA5无法识别[31]。据此认为,在病毒复制早期,NS3虽未能有效切割MAVS介导天然免疫逃逸,但此囊泡结构阻止病毒复制过程中产生的危险信号被天然免疫识别分子识别;在病毒复制晚期,复制复合体的代谢可能导致复制产物等危险信号的释放,而病毒复制产物被MDA5识别,诱生干扰素,但此时复制累积所产生的NS3蛋白通过反式作用有效切割MAVS,进而阻止干扰素产生。
综上所述,本研究提出了一个HCV NS3/4A切割MAVS使病毒逃逸宿主天然免疫的模型:HCV NS3/4A切割MAVS只依赖NS3表达水平,不依赖其存在形式和基因型;NS3/4A切割MAVS对病毒逃逸干扰素的作用仅局限于病毒复制晚期,对病毒感染早期建立复制稳态无显著影响。
[1] Rosen HR. Clinical practice. Chronic hepatitis C infection [J]. N Engl J Med, 2011, 364(25): 2429-2438.
[2] Tellinghuisen TL, Evans MJ, Von Hahn T, You S, Rice CM. Studying hepatitis C virus: making the best of a bad virus [J]. J Virol, 2007, 81(17): 8853-8867.
[3] Bowen DG, Walker CM. The origin of quasispecies: cause or consequence of chronic hepatitis C viral infection? [J]. J Hepatol, 2005, 42(3): 408-417.
[4] Chisari FV. Unscrambling hepatitis C virus-host interactions [J]. Nature, 2005, 436(7053): 930-932.
[5] Thomas MG, Loschi M, Desbats MA, Boccaccio GL. RNA granules: the good, the bad and the ugly [J]. Cell Signal, 2011, 23(2): 324-334.
[6] Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence [J]. Hepatology, 2013, 57(4): 1333-1342.
[7] Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus [J]. Nat Rev Microbiol, 2007, 5(6): 453-463.
[8] Nguyen H, Sankaran S, Dandekar S. Hepatitis C virus core protein induces expression of genes regulating immune evasion and anti-apoptosis in hepatocytes [J]. Virology, 2006, 354(1): 58-68.
[9] Horner SM. Insights into antiviral innate immunity revealed by studying hepatitis C virus [J]. Cytokine, 2015, 74(2): 190-197.
[10] Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity [J]. Proc Natl Acad Sci USA, 2005, 102(49): 17717-17722.
[11] Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, Rice CM, Dustin LB. Hepatitis C virus induces interferon-λ and interferon-stimulated genes in primary liver cultures [J]. Hepatology, 2011, 54(6): 1913-1923.
[12] Israelow B, Narbus CM, Sourisseau M, Evans MJ. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection [J]. Hepatology, 2014, 60(4): 1170-1179.
[13] Binder M, Kochs G, Bartenschlager R, Lohmann V. Hepatitis C virus escape from the interferon regulatory factor 3 pathway by a passive and active evasion strategy [J]. Hepatology, 2007, 46(5): 1365-1374.
[14] Yi Z, Fang C, Zou J, Xu J, Song W, Du X, Pan T, Lu H, Yuan Z. Affinity purification of the hepatitis C virus replicase identifies valosin-containing protein, a member of the ATPases associated with diverse cellular activities family, as an active virus replication modulator [J]. J Virol, 2016, 90(21): 9953-9966.
[15] Yi Z, Pan T, Wu X, Song W, Wang S, Xu Y, Rice CM, Macdonald MR, Yuan Z. Hepatitis C virus co-opts Ras-GTPase-activating protein-binding protein 1 for its genome replication [J]. J Virol, 2011, 85(14): 6996-7004.
[16] Shiina M, Rehermann B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function [J]. Hepatology, 2008, 47(2): 385-395.
[17] Yi Z, Yuan Z, Rice CM, Macdonald MR. Flavivirus replication complex assembly revealed by DNAJC14 functional mapping [J]. J Virol, 2012, 86(21): 11815-11832.
[18] Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, Mckeating JA, Rice CM. Complete replication of hepatitis C virus in cell culture [J]. Science, 2005, 309(5734): 623-626.
[19] Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro [J]. Proc Natl Acad Sci USA, 2005, 102(26): 9294-9299.
[20] Jones CT, Catanese MT, Law LM, Khetani SR, Syder AJ, Ploss A, Oh TS, Schoggins JW, Macdonald MR, Bhatia SN, Rice CM. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system [J]. Nat Biotechnol, 2010, 28(2): 167-171.
[21] Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED, Rice CM, Dustin LB. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells [J]. Hepatology, 2008, 48(6): 1843-1850.
[22] Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA [J]. Nature, 2008, 454(7203): 523-527.
[23] Uzri D, Gehrke L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities [J]. J Virol, 2009, 83(9): 4174-4184.
[24] Cao X, Ding Q, Lu J, Tao W, Huang B, Zhao Y, Niu J, Liu YJ, Zhong J. MDA5 plays a critical role in interferon response during hepatitis C virus infection [J]. J Hepatol, 2015, 62(4): 771-778.
[25] Du X, Pan T, Xu J, Zhang Y, Song W, Yi Z, Yuan Z. Hepatitis C virus replicative double-stranded RNA is a potent interferon inducer that triggers interferon production through MDA5 [J]. J Gen Virol, 2016, 97(11): 2868-2882.
[26] Ahlquist P. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses [J]. Nat Rev Microbiol, 2006, 4(5): 371-382.
[27] Denison MR. Seeking membranes: positive-strand RNA virus replication complexes [J]. PLoS Biol, 2008, 6(10): e270.
[28] Miller S, Sparacio S, Bartenschlager R. Subcellular localization and membrane topology of the Dengue virus type 2 non-structural protein 4B [J]. J Biol Chem, 2006, 281(13): 8854-8863.
[29] Mackenzie J. Wrapping things up about virus RNA replication [J]. Traffic, 2005, 6(11): 967-977.
[30] Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. Virus factories: associations of cell organelles for viral replication and morphogenesis [J]. Biol Cell, 2005, 97(2): 147-172.
[31] Neufeldt CJ, Joyce MA, Van Buuren N, Levin A, Kirkegaard K, Gale M Jr, Tyrrell DL, Wozniak RW. The hepatitis C virus-induced membranous web and associated nuclear transport machinery limit access of pattern recognition receptors to viral replication sites [J]. PLoS Pathog, 2016, 12(2): e1005428.
s. YUAN Zhenghong, E-mail: zhyuan@shaphc.org; YI Zhigang, E-mail: zgyi@fudan.edu.cn
KineticsofhepatitisCvirusNS3/4A-mediatedMAVScleavageduringhepatitisCvirusinfection
XU Jun, DU Xiaoting, ZHANG Yang, YUAN Zhenghong, YI Zhigang
DepartmentofMedicalMicrobiologyandParasitology,KeyLaboratoryofMedicalMolecularVirologyofMinistriesofEducationandHealth,SchoolofBasicMedicalSciences,FudanUniversity,Shanghai200032,China
Hepatitis C virus (HCV) protease NS3/4A cleaves mitochondrial antiviral signaling protein (MAVS) to evade host innate recognition and interferon production. However, HCV infection induces interferon production in certain cell types and HCV could replicate in cells expressing NS3/4A cleavage-resistant MAVS. The roles of NS3/4A-mediated MAVS cleavage in host innate evasion and restriction of viral replication need to be clarified. In this study, by using an HCV NS3/4A-mediated MAVS cleavage reporter system, in which Huh7.5 cells express GFP-NLS-MAVS-TM462, we dissected the kinetics of MAVS cleavage during HCV infection. It was found that MAVS was barely cleaved during the early time but was efficiently cleaved only in the late time during viral infection. Using ypet-tagged HCV virus and Huh7.5 cells expressing RFP-NLS-MAVS-TM462, we dissected the MAVS cleavage in the HCV-positive (ypet-positive) cells on a single-cell manner. It was found that MAVS cleavage occurred only in part of the HCV-infected cells. We also assessed the MAVS cleavage by HCV NS3/4A from different genotypes, and found the cleavage efficiency was correlated with the protein level of HCV NS3/4A. Taken together, we found NS3/4A-mediated MAVS cleavage occurs in the late stage during viral replication and is correlated with NS3 protein level. We proposed HCV NS3/4A-mediated MAVS cleavage does not contribute to host innate evasion during early viral replication step.
Hepatitis C virus; Nonstructural protein 3/4A; Mitochondrial antiviral signaling protein; Interferon
国家重点基础研究发展计划(2015CB554301、2012CB519005),中德跨学科重大合作研究项目(81461130019)
袁正宏,易志刚
2017-05-18)
猜你喜欢
广西医学(2022年22期)2023-01-30
野生动物学报(2020年1期)2020-02-21
文苑(2018年22期)2018-11-19
中国高原医学与生物学杂志(2017年4期)2017-03-08
中国组织化学与细胞化学杂志(2016年3期)2016-02-27
中国粮油学报(2016年5期)2016-01-23
西南军医(2016年6期)2016-01-23
中国继续医学教育(2015年6期)2016-01-07
中国医疗美容(2015年2期)2015-07-19
中国生化药物杂志(2015年4期)2015-07-07
