
安痛舒贴质量标准研究
2017-09-28 11:34刘春河张国跃
中国药业 2017年18期
刘春河,张国跃
(1.武汉科技大学附属天佑医院,湖北 武汉 430064; 2.陕西省食品药品检验所,陕西 西安 710065)
安痛舒贴质量标准研究
刘春河1,张国跃2
(1.武汉科技大学附属天佑医院,湖北 武汉 430064; 2.陕西省食品药品检验所,陕西 西安 710065)
目的建立安痛舒贴的质量标准。方法 采用薄层色谱法定性鉴别姜黄、甘遂,采用高效液相色谱法测定马钱子和当归含量。马钱子含量测定,Ultimate C18色谱柱、Capcell Pak C18MG柱(250 mm×4.6mm,5 m),乙腈 -0.01mol/L磷酸二氢钾与0.02 mol/L庚烷磺酸钠等量混合溶液(用磷酸调pH至2.8)(20∶80)为流动相,流速为1.0 mL/min,柱温为35℃,检测波长为260 nm。当归含量测定,色谱柱同上,甲醇 -0.05mol/L磷酸溶液(26∶74)为流动相,检测波长为319 nm,柱温为35℃。结果 士的宁进样量在0.024 92~0.438 4 g、马钱子碱进样量在0.023 92~0.478 4 g、阿魏酸进样量在0.089 2~0.802 8 g范围内,与峰面积线性关系良好;回归方程分别为Y=5.598 24×106X+77 520(r=0.999 9)、Y=6×10-7X+0.007 5(r=0.999 8)、Y=6×10-2X+0.563 87(r=0.999 8),平均加样回收率马钱子碱为99.97%,士的宁为96.87%,阿魏酸为97.68%(n=6)。结论 该质量标准操作简便易行,定性、定量方法准确度高、重复性好,检测设定指标合理,可用于安痛舒贴的质量控制。
薄层色谱法;高效液相色谱法;安痛舒贴;桃仁;当归;甘遂;士的宁;马钱子碱;阿魏酸;含量测定;定性鉴别;质量控制
Abstract:Ob jective To establish the quality standard for the Antongshu Plaster.M ethods TLC was used for the qualitative identification of Curcuma longa,Euphorbia kansui,HPLC was used for the content determination of Strychnos nux-vomica and Angelica sinensis.The content determination of Strychnos nux-vomica:Ultimate C18column and Capcell Pak C18MG column(250 mm×4.6 mm,5μm)were adopted with acetonitrile -0.01 mol/L KH2PO4and 0.02 mol/L sodium heptanesulfonate mixture solution(pH=2.8 adjusted with phosphoric acid) (20∶80)as the mobile phase,the flow rate of 1.0 m L/min,the column temperature was 35℃ ,the detection wavelength was 260 nm.The content determination of Angelica sinensis:The column was the same as above,the mobile phase was methanol- 0.05 mol/L phosphoric acid solution(26∶74),the detection wavelength was 319 nm,the column temperature was 35℃.Results The strychnine,brucine and ferulic acid showed a good linear in the range of 0.024 92-0.438 4μg,0.023 92-0.478 4μg,0.089 2-0.802 8μg,respectively,and the regression equations were Y=5.598 24×106X+77 520(r=0.999 9),Y=6×10-7X+0.007 5(r=0.999 8),Y=6×10-2X+0.563 87(r=0.999 8),respectively.The average recovery rate of brucine,strychnine and ferulic acid were 99.97%,96.87% and 97.68%,respectively.Conclusion The quality standard is simple and easy to operate.The qualitative and quantitative methods have high accuracy,good repeatability and reasonable test setting index,which can be used for the quality control of Antongshu Plaster.
Key words:TLC;HPLC;Antongshu Plaster;Prunus persica;Angelica sinensis;Euphorbia kansui;strychnine;brucine;ferulic acid;content determination;qualitative identification;quality control
安痛舒贴系我军某大型综合医院依据明代医家王肯堂《六科准绳》记载的“阿魏化痞膏”,结合临床经验,按照传统工艺研制的外用膏剂,由当归、草乌、马钱子、姜黄、大黄、乳香、没药、麝香、冰片等15余味药材制成,具有清热解毒、消肿散结、活血止痛、逐水利尿等功效,适用于肩周炎及扭伤、肿瘤等各种原因引起的疼痛。其质量标准收载于“兰制字中(2011)F68122”中,原标准仅有大黄、当归和香附的薄层色谱鉴别法,马钱子等毒性药材没有定量或限度检查,也没有君药的定量检测,制剂的有效性和安全性不能得到有效控制。受该医院的委托,参照国家药典委员会关于药品质量标准技术要求,验证了原质量标准中的薄层色谱鉴别法并进行了补充和修订,建立了姜黄、甘遂的薄层色谱鉴别法和桃仁的显微鉴别法,建立了薄层色谱法检查乌头碱限度的方法,起草了高效液相色谱法测定马钱子含量(以士的宁、马钱子碱为定量指标)和当归含量(以阿魏酸为定量指标)的方法,并进行了方法学验证。修订后的质量标准方法简便易行、准确、重复性较好,可为该制剂的质量控制提供监测手段。现报道如下。
1 仪器与试药
1.1 仪器
BX41型显微镜(奥林巴 斯公司);Sartorius AGME235s型分析天平(赛多利斯公司);UV-2550型紫外分光光度计(日本岛津公司);1260型高效液相色谱仪(安捷伦公司)、LC30AD型高效液相色谱仪(日本岛津公司)和 Waters 2695-2487型高效液相色谱仪(Waters公司),Waters Empower工作站;Ultimate C18柱及 Capcell Pak C18MG色谱柱(250 mm×4.6 mm,5μm);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药
试验用样品安痛舒贴(批号分别为 150101,150202,150303),不含姜黄、甘遂、桃仁、大黄、香附、马钱子、当归等的阴性样品,马钱子、当归等药材粉末,均由委托方提供;对照药材和对照品均由中国食品药品检定研究院提供,包括大黄(批号120902-201010)、当归(批号120927-201014)、甘遂(批号1042-9902)、姜黄(批号1188-200101)、姜黄素(批号0823-9802)、士的宁(批号110705-200306)、马钱子碱(批号:110706-200505)、阿魏酸(批号110773-200611)。乙腈、甲醇为色谱纯,其他试剂均为分析纯,水为超纯水。薄层预制板(青岛海洋化工有限公司),自制薄层板。
2 方法与结果
2.1 桃仁的显微鉴别
取本品粉末,置显微镜下观察:石细胞黄色或黄棕色,侧面观呈贝壳形、盔帽形、弓形或椭圆形,高54~153μm,底部宽约180μm,壁一边较厚,层纹细密;表面观呈类圆形、圆多角形或类方形,底部壁上纹孔大而较密,符合2015年版《中国药典(一部)》“桃仁”的显微特征[1],列入质量标准。详见图1。

图1 桃仁显微特征图
2.2 姜黄的薄层色谱鉴别
取本品1贴,刮取药膏,加无水乙醇25 m L,振摇30min,再放置30min,滤过,滤液蒸干,残渣加无水乙醇5mL使溶解,作为供试品溶液。取不含姜黄的阴性样品,同法制成姜黄阴性对照品溶液。另取姜黄对照药材0.5 g,同法制成对照药材溶液。再取姜黄素对照品,加无水乙醇制成每1mL含0.5mg的溶液,作为对照品溶液。分别吸取上述4种溶液各4μL,点于同一硅胶G薄层板上,以三氯甲烷 -甲醇 -甲酸(96∶4∶0.7)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液和对照品溶液色谱相应位置上,显相同颜色的荧光斑点,阴性对照无干扰,斑点分离较好,方法可行。详见图2。

图2 姜黄薄层色谱鉴别图
2.3 甘遂的薄层色谱鉴别
取本品2贴,刮取药膏,加乙醚25 mL,超声处理30min,滤过,弃滤液,药渣挥干溶剂,加乙醇25m L,超声处理40min,滤过,滤液蒸干,残渣加乙醇1mL使溶解,作为供试品溶液。取不含甘遂的阴性样品,同法制成甘遂阴性对照品溶液。取甘遂对照药材1 g,加甲醇25 mL,超声处理40min,滤过,滤液蒸干,残渣加乙醇1mL使溶解,作为对照药材溶液。分别吸取上述3种溶液各2μL,点于同一硅胶G薄层板上,以石油醚(30~60℃)-丙酮(5∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,置 105℃加热至斑点显色清晰。置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液色谱相应位置上,显相同颜色的荧光主斑点,斑点分离度尚好,阴性无干扰,详见图3。

图3 甘遂薄层色谱鉴别图
2.4 马钱子含量测定
2.4.1 色谱条件
色谱柱为Ultimate C18柱、Capcell Pak C18MG柱(250mm×4.6mm,5μm),以十八烷基硅烷键合硅胶为填充剂;乙腈-0.01 mol/L磷酸二氢钾与0.02 mol/L庚烷磺酸钠等量混合溶液(用磷酸调pH至2.8)(20∶80)为流动相;检测波长为260 nm;柱温为35℃;理论板数按士的宁峰计算应不低于6 000。
2.4.2 溶液制备
对照品溶液:取士的宁对照品6 mg、马钱子碱对照品5 mg,精密称定,分别置10 m L容量瓶中,加三氯甲烷使溶解,并稀释至刻度,摇匀。分别精密量取1mL,置同一25mL容量瓶中,用甲醇稀释至刻度,摇匀,即得(每1m L含士的宁0.024mg、马钱子碱0.02mg)。
供试品溶液:取本品5贴,刮取药膏,混匀,取约5 g,精密称定,置具塞锥形瓶中,加氢氧化钠试液5 m L,振摇,放置30 min,精密加三氯甲烷25 m L,称定质量,超声处理(功率500W,频率40 kHz)50min,取出,放冷,再称定质量,用三氯甲烷补足减失的质量,摇匀,分取三氯甲烷液,用铺有少量无水硫酸钠的滤纸滤过,弃去初滤液,精密量取续滤液10 mL,蒸干,残渣加甲醇使溶解,并转移至10mL容量瓶中,加甲醇至刻度,摇匀,微孔滤膜(0.45μm)滤过,取续滤液,即得。
2.4.3 测定波长选择
取士的宁及马钱子碱混合对照品溶液(约各含25μg/m L),在200~400nm波长处进行光谱扫描。结果士的宁、马钱子碱混合对照品溶液的最大吸收波长在258.8 nm,与2015年版《中国药典(一部)》“马钱子”药材中士的宁、马钱子碱测定波长相近,故拟订测定波长为260 nm。
2.4.4 方法学考察
专属性试验:取不含马钱子的阴性样品,按供试品溶液制备方法制备阴性对照品溶液,进样测定。结果显示,阴性对照品溶液对测定无干扰,专属好,见图4。
线性关系考察:分别精密吸取士的宁、马钱子碱混合对照品溶液(24.92μg/m L和23.92μg/m L)1,2,5,10,15,20μL,测定,以进样量(X)为横坐标、峰面积(Y)为纵坐标,进行线性回归,得回归方程分别为 Y=5.598 24×106X+775 20(r=0.999 9),Y=6×10-7X+0.007 5(r=0.999 8)。结果表明,士的宁进样量在0.024 92~0.438 4μg范围内、马钱子碱进样量在0.023 92~0.478 4μg范围内与峰面积线性关系良好。
稳定性试验:取同一供试品溶液,分别在0,1,5,9,12 h时进样测定。结果的 RSD为1.21%(n=5),表明供试品溶液在12 h内稳定性较好。
精密度试验:取士的宁、马钱子碱混合对照品溶液,重复进样6次,结果士的宁、马钱子碱峰面积积分值的 RSD分别为0.45%和0.53%(n=6),表明仪器精密度良好。

图4 马钱子含量测定高效液相色谱图
重复性试验:精密称取同一样品共6份,依法制备供试品溶液,测定。结果马钱子碱、士的宁的 RSD分别为2.99%和2.78%(n=6),表明方法重复性好。
中间精密度试验:按拟订试验方法和色谱条件,由本实验室的其他试验员测定马钱子碱和士的宁的含量。试验结果基本相同,RSD分别为2.78%和3.02%。
加样回收试验:精密称取样品2.5 g共6份,分别精密加入含有马钱子碱0.301 5 g/L和士的宁0.180 5 g/L的混合对照品溶液1mL,依法制备供试品溶液,测定。结果见表1。
耐用性试验:分别通过改变柱温、流速和色谱柱来考察方法耐用性,结果除保留时间有轻微提前或拖后外,峰形、峰面积及与其他色谱峰分离程度均无明显变化,耐用性较好。
2.4.5 样品含量测定
取样品适量,依法制备供试品溶液,测定其他样品中马钱子碱、士的宁含量,结果见表2。
2.5 当归含量测定
2.5.1 色谱条件
色谱柱:同 2.4.1项;流动相:甲醇 -0.05 mol/L磷酸溶液(26∶74);检测波长:319 nm;以十八烷基硅烷键合硅胶为填充剂;理论板数按阿魏酸峰计算应不低于3 000。
2.5.2 溶液制备
对照品溶液:取阿魏酸对照品适量,精密称定,加甲醇制成每1mL含16μg的溶液,即得。

表1 马钱子碱及士的宁加样回收试验(n=6)

表2 样品含量测定结果( g/g)
供试品溶液:取重量差异项下本品,混匀,取约2.5 g,精密称定,加硅藻土2 g,研匀,将其转移至具塞锥形瓶中,研钵用50m L碱性甲醇(用氢氧化钠试液调pH至10~11)分次洗涤,洗涤液转入锥形瓶中,超声处理(功率500W,频率40 kHz)1 h,滤过,滤液蒸干,残渣加水20 mL使溶解,用盐酸试液调 pH至2,用乙醚提取4次,每次30m L,合并乙醚液,挥干,残渣加甲醇溶解,并转移至10m L容量瓶中,用甲醇稀释至刻度,即得。
2.5.3 测定波长选择
取阿魏酸对照品溶液,以甲醇溶液为空白,在250~350 nm波长范围扫描,阿魏酸的最大吸收波长为319 nm。见图5。

图5 阿魏酸紫外光扫描图
2.5.4 方法学考察
专属性试验:取不含当归的阴性样品,按2.5.2项下供试品溶液制备方法制备阴性对照品溶液,按拟订色谱条件,将对照品溶液、供试品溶液、阴性对照品溶液注入高效液相色谱仪,测定。结果阴性对照品溶液在与对照品溶液相同保留时间处无吸收峰。详见图6。
线性关系试验:取质量浓度分别为17.84μg/mL和53.52μg/m L的阿魏酸对照品溶液,按拟订色谱条件分别进样5,10μL和5,10,15μL,记录色谱图,计算峰面积,以峰面积与进样量进行线性回归,得回归方程Y=6×10-2X+0.563 87,r=0.999 8(n=6)。结果表明,阿魏酸进样量在0.089 2~0.802 8μg范围内与峰面积线性关系良好。
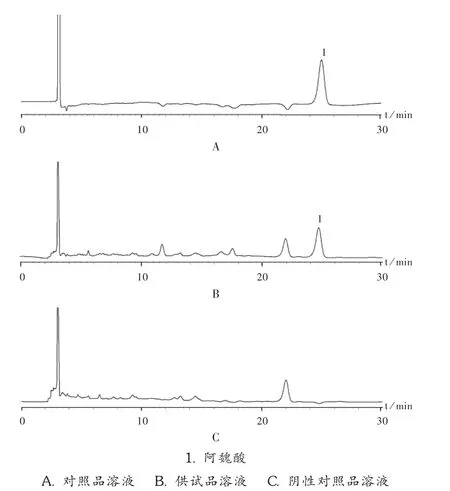
图6 当归含量测定高效液相色谱图
精密度试验:分别精密吸取同一对照品溶液10μL,注入高效液相色谱仪,连续进样6次,测定峰面积。结果的 RSD为0.79%(n=6),表明仪器精密度良好。
稳定性试验:精密吸取同一供试品溶液10μL,分别在0,2,4,6,12,24 h时注入高效液相色谱仪,测定峰面积。结果的 RSD为1.21%(n=6),表明阿魏酸在24 h内稳定性良好。
重复性试验:取同一样品,平行制备6份供试品溶液,依法进样,测定阿魏酸的含量。结果的 RSD为1.89%(n=6)。表明本方法重复性良好。
加样回收试验:在具塞锥形瓶中精密加入阿魏酸对照品溶液1 m L(质量浓度为0.253 5 g/L),挥干溶剂,取已知含量的安痛舒贴样品适量,混匀,取约1.5 g,精密称定,加硅藻土2 g,研匀,依法平行制备供试品溶液6份,测定,计算回收率。结果见表3。

表3 阿魏酸加样回收试验(n=6)
2.5.5 样品含量测定
取3批安痛舒贴样品,依法制备供试品溶液,测定含量,结果见表4。

表4 3批样品测定结果
3 讨论
据报道,姜黄的薄层色谱鉴别可用三氯甲烷-甲醇(95∶5)作为展开剂[2-3],但经试验后发现,分离效果不好,斑点扩散严重。曹智勇等[4]报道,用苯-丙酮作为展开剂进行甘遂的薄层色谱鉴别,因苯的毒性比较大,弃用,改用文中的展开剂效果尚好。上述姜黄、甘遂薄层色谱鉴别方法在制订时曾通过改变试验条件,如不同薄层板(自制和预售)、不同温湿度、点样量和展距等来考察耐用性,结果斑点的分离度、比移值(Rf)等均符合要求,表明该方法的耐用性较好。
马钱子为马钱科植物马钱 nux′vomica L.的干燥成熟种子,主要成分为士的宁和马钱子碱,另外还含番木鳖次碱、番木鳖苷、γ-及β-可鲁勃林、马钱子新碱等,口服时治疗量与中毒量接近[5]。马钱子外用的毒性考察报道少见,但也需在质量标准中加以控制。用高效液色谱法测定制剂中士的宁、马钱子碱的含量较常见,本研究中参考了相关报道[6-9],结合处方和制备工艺,确定了文中的测定方法。另外,根据安痛舒贴的处方和制备工艺,分别考察了不同的处理方法,如加热回流2 h或1 h,超声处理30min或60min,结果显示,用不同处理方法处理样品,士的宁、马钱子碱的总含量无显著差异,而超声处理的方法简便、快捷,故最终确定了文中供试品溶液制备方法。
当归为伞形科植物当归 Angelica sinensis(Oliv.)Diels的干燥根,有着广泛的药理活性,为医家常用,常有“十方九归”之说。现代研究表明,当归对血液及造血系统、抗炎及免疫系统、中枢神经系统等均有作用,对缺血损伤具有保护作用,且有较强的镇痛作用[9-14]。本试验中用高效液相色谱法测定当归中的重要成分阿魏酸,曾用文献报道的0.2%甲醇乙腈 -0.1%磷酸0.1%三乙胺水溶液(20∶80)作为流动相,但阿魏酸的出峰时间较早,其他色谱峰有干扰;流动相为甲醇-1%冰醋酸 -甲氢呋喃(30∶70∶1)时,阿魏酸的色谱峰有拖尾现象[15-17]。采用本研究中所用流动相出峰时间适宜,峰形较好,分离度能达到要求。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科学出版社,2015:277-278.
[2]李 明,周大颖,田 园,等.中药姜黄的薄层色谱研究[J].贵州化工,2008,33(1):31-34.
[3]陈玲珑,田其学,吴劲松.复方消炎散中冰片和姜黄的薄层色谱鉴别[J].湖南中医杂志,2013,29(6):124.
[4]曹智勇,虞金宝.冻疮涂膜剂的质量标准研究[J].江西中医学院学报,2003,15(3):48-50.
[5]张德放,邱维彬,王 坤.马钱子的毒性及中毒解救的研究[J].辽宁中医杂志,2011,35(4):713-715.
[6]吴春燕,贾建忠,李小安,等.HPLC法同时测定腰腿痛丸中麻黄碱、士的宁及马钱子碱的含量[J].西北药学杂志,2014,29(4):370-372.
[7]那 微,张清波,张淑华,等.HPLC法测定骨筋丸系列品种中马钱子碱和士的宁[J].中成药,2012,34(4):759-762.
[8]潘金火,严国俊,周娟娟.马钱子粉中士的宁和马钱子碱含量测定方法的研究[J].中国药学杂志,2010,45(13):1024-1029.
[9]周 玲,孙汉斌,邓旭坤,等.高效液相色谱法测定3种马钱属药材中士的宁和马钱子碱的含量[J].中国医院药学杂志,2012,32(16):1301-1302.
[10]刘如秀,刘 宇,汪艳丽,等.当归的药理作用[J].西部中医药,2014,27(11):153-156.
[11]田景祥.当归的有效成分及其药理作用的研究进展[J].工企医刊,2013,26(4):364-365.
[12]李 曦,张丽宏,王晓晓,等.当归化学成分及药理作用研究进展[J].中药材,2013,36(6):1023-1028.
[13]宋 敏,黎七雄,何成龙.当归提取物的鉴定及镇痛活性观察[J].咸宁学院学报(医学版),2009,23(3):194-196.
[14]杨 瑜,查仲玲,朱 蕙,等.当归提取物的镇痛作用[J].医药导报,2002,21(8):481-482.
[15]李成义,王延惠,魏学明,等.HPLC测定不同产地当归中阿魏酸的含量[J].西部中医药,2012,25(1):34-36.
[16]陈超超,王 艳,梁 超.高效液相色谱法测定当归中阿魏酸的含量[J].成都大学学报(自然科学版),2008,27(4):284-286.
[17]刘蓉梅,黄罗生.高效液相色谱法测定当归中阿魏酸含量方法的探讨[J].东南大学学报(医学版),2003,22(2):98-101.
Quality Standard of Antongshu Plaster
Liu Chunhe1,Zhang Guoyue2
(1.Tianyou Hospital Affiliated to Wuhan University of Science and Technology,Wuhan,Hubei,China 430064; 2.Shaanxi Institute for Food and Drug Control,Xi′an,Shaanxi,China 710065)
R284.1;R289.6
A
1006-4931(2017)18-0017-05
10.3969/j.issn.1006-4931.2017.18.006
2017-05-09)
刘春河,男,主管药师,主要从事临床药品检验工作,(电子信箱)2047769380@qq.com。
猜你喜欢
化工管理(2022年14期)2022-12-02
Digital Chinese Medicine(2022年2期)2022-07-02
九江学院学报(自然科学版)(2022年2期)2022-07-02
中国化肥信息(2022年3期)2022-05-05
汽车实用技术(2022年4期)2022-03-07
口腔护理用品工业(2021年4期)2021-11-02
健康之家(2021年19期)2021-05-23
中国药学药品知识仓库(2021年18期)2021-02-28
国际放射医学核医学杂志(2021年10期)2021-02-28
Medical Data Mining(2019年2期)2019-07-16
