
先天性隐眼遗传致病基因研究现状
2017-09-19 02:20张夏茵王婧荟龙尔平吴晓航李王婷王东妮林浩添
转化医学电子杂志 2017年8期
张夏茵,王婧荟,龙尔平,吴晓航,李王婷,王东妮,林浩添
(中山大学中山眼科中心,眼科学国家重点实验室,广东广州510060)
·专家述评·
先天性隐眼遗传致病基因研究现状
张夏茵,王婧荟,龙尔平,吴晓航,李王婷,王东妮,林浩添
(中山大学中山眼科中心,眼科学国家重点实验室,广东广州510060)
先天性隐眼是一种终生致盲致畸的罕见眼病,发病机制至今未完全明确.患儿出生即表现双眼睑缺失,眼眶表面完全被连续性皮肤覆盖,眼眶内仅有眼球遗迹或完全无眼球.先天性隐眼与遗传致病基因异常密切相关,目前已发现三个隐眼遗传致病基因FRAS1,FREM2,GRIP1分别定位于4号、13号和12号染色体的长臂上.本文回顾近年国内外先天性隐眼的病例报告及遗传学研究成果,对以上三个遗传致病基因的位置结构、功能及突变的研究现状进行总结,为隐眼遗传学发病机制的进一步研究及其在产前诊断中的转化应用提供参考.
先天性隐眼;隐眼畸形综合症;遗传致病基因; FRAS1;FREM2;GRIP1
0 引言
先天性隐眼(congenital cryptophtalmos)是一种终生致盲致畸的罕见眼病,又称无睑症,由Zehender于1872年首次报道,患病率为3/100 000~14/100 000[1-2].一般为常染色体隐性遗传,可单眼或双眼发病[3].隐眼患者表现为眼球和眼睑部位先天畸形,眼球常被连续性的皮肤所遮盖而无睑裂,仅有眼球遗迹或完全无眼球.眼球前部多存在一个泡形结构,内有晶体的残留和玻璃体,眼球后部几乎正常.患者眼窝可触摸到皮下有一定活动度的球形物.有些病例在强光刺激时可见到因眼轮匝肌反射性收缩造成的皮肤皱缩,并对光源有一定的跟随运动,提示这些患者可能尚有光感[4-6].然而,因为得不到光的刺激,患儿的光感逐步衰退直至完全丧失.隐眼虽可以是一个独立的临床表型,但常作为隐眼畸形综合症(fraser syndrome,FS;OMIM 219000)的各种临床表型之一出现[7].FS由Fraser在1962年首次描述并命名[8],是一种罕见的常染色体隐性遗传疾病,家族性畸形多见[9].表现为隐眼合并多种其它先天异常,如皮肤性并指(趾)畸形、泌尿生殖器畸形、鼻畸形、牙齿畸形、腭裂、面部和眶骨发育不全、耳聋、脑膜膨出、智力迟钝等[10-11].
随着基因测序成本的下降和临床筛查及转化应用研究的普及推广,目前已经证实隐眼的发生与遗传致病基因突变如4号、13号以及12号染色体长臂上的基因FRAS1,FREM2,GRIP1异常有关[8,12-14].近年来,国外许多学者对以上三种基因进行了结构和功能探索,但多数仅限于临床病例报道而尚未有遗传学的基因发病机制研究.本文将对隐眼遗传致病基因的位置结构、功能及突变的研究现状进行综述,为明确隐眼的发病机制、推动临床转化应用,并最终杜绝这类重大的先天遗传缺陷患儿的出生奠定重要基础.
1 基因位置、结构
目前,已证实的FS致病基因按照发现的时间顺序分别是4号、13号以及12号染色体长臂上的基因FRAS1,FREM2,GRIP1,其中FRAS1基因是FS的主要致病基因,检测出FREM2和GRIP1异常的比例大致相等.
2003年McGregor在6家系14位患者中发现FRAS1基因突变,通过对基因库的BLAST分析确定患者FRAS1基因的外显子突变情况[11].FRAS1基因位于染色体4q21.21上,由74个外显子组成,与编码细胞外基质蛋白的基因存在序列相似性[15].在胚胎和成人的多种上皮组织以及肾脏、胰腺和丘脑中均有表达.
2005年Jadeja等[16]在FS动物模型——小鼠my (myelencephalic blebs)品系中发现 FREM2基因突变,并在2家系2位FS患者中通过直接测序FREM2基因外显子发现了核苷酸 5914G>A(H)突变.FREM2基因位于染色体13q13.3上,由24个外显子组成,其中外显子4编码了超过一半的氨基酸.相比表皮发育过程中FRAS1基因在表皮的高度表达,FREM2基因在滤泡间区的表达量较少,但在内胚层、外胚层和一系列重要的神经信号中心(包括背侧前脑的中线、中脑和后脑边界)有动态表达.
2012年Vogel等[17]基于细胞分子水平及GRIP1基因敲除小鼠模型的前期研究,第一次在无FRAS1和FREM2基因异常的2家系2位FS患儿血中检测出GRIP1基因突变.患者GRIP1基因外显子结构异常直接导致转录异常和翻译蛋白缩短,从而影响Grip1蛋白生物功能引发FS表型.GRIP1基因位于染色体12q14.3上,由24个外显子组成,编码含7个PDZ(postsynaptic density-95/discs large/zona occludens-1)结构域的胞质支架蛋白,连接Fras1蛋白和Frem2蛋白的C-末端残基[8].在胚胎早期发育过程中GRIP1基因表达与细胞和细胞外基质间相互作用密切相关.GRIP1基因在发育中的神经系统等多区域表达,也与其在调节蛋白质转运过程中扮演的多种角色有关.
目前发现还有一部分先天性隐眼与 FRAS1,FREM2,GRIP1的突变无关,推测仍有其他的可导致隐眼发病的致病基因存在.2009年Carney等[18]在FS动物模型-斑马鱼鱼鳍突变体中利用候选基因预测与测序分析、定位克隆等技术发现 HMCN1和FBN2基因异常,猜测它们可能是FS的重要候选基因.VWA2基因也可能与隐眼发生相关,Richardson等[19]在2013年发现FRAS1基因表达异常的斑马鱼和小鼠模型中均缺失了可与Fras1直接作用的AMACO (matrilins and collagens)蛋白,AMACO蛋白由VWA2基因编码,阻碍VWA2基因表达导致FRAS1突变斑马鱼的表型更加严重.但以上HMCN1、FBN2和VWA2基因变异在隐眼患者中尚未得到证实.
2 基因的功能
FRAS1,FREM2,GRIP1三种基因功能目前均已在分子层面明确,得到了小鼠活体模型的验证,并在FS患者中发现基因异常可导致出现经典的隐眼表型[17].它们通过表达Fras1,Frem2和Grip1三种蛋白影响组织的结构完整和器官发育过程中上皮和间质间的相互联系.
Fras1蛋白与Frem2蛋白同为位于细胞外基质(extracellular matrix,ECM)的多域跨膜蛋白,是致密层下区的重要组成成分,均有连续硫酸软骨素蛋白多糖(chondroitin sulfate proteoglycan,CSPG)模体和多个CalX-β结构域(图1),与具有细胞粘附功能的跨膜蛋白——钙粘素的结构相像.CSPG模体可结合钙离子、bFGF以及PDGF-AA,并发生构象转变,其中与钙离子的结合对于FREM2基因发挥功能是必需的[11,16],而CalX-β结构域的功能尚不清楚.
Fras1与Frem2两种蛋白在细胞外基质的结构形成和信号传递网络中发挥着重要作用[11].Kiyozumi等[20]发现Frem2作为中间蛋白可与Fras1,Frem1相互作用形成稳定的Fras1/Frem蛋白复合物定位在基底膜上.Fras1蛋白与Frem2蛋白不仅是ECM的构成成分,同时对细胞外环境中各生长因子的活动起调控作用.胶原Ⅶ是致密层下一种主要的锚定成分,Fras1蛋白与Frem2蛋白对于仅在胚胎发育晚期增多的胶原Ⅶ起补偿作用[14],也与胶原Ⅳ、Ⅴ、Ⅵ、碱性成纤维细胞生长因子、血小板源性生长因子间作用密切[8].ECM蛋白通过彼此间的相互协作主要调节细胞的迁移、增殖、分化以及细胞间信号转导等,这些功能在胚胎发育的过程中非常重要[21].Fras1/Frem蛋白复合物与胚胎期上皮和间充质细胞间的相互作用密切相关,动物实验表明缺少Frem2蛋白会破坏复合物形成,影响表皮粘连以及表皮基底膜和下部真皮结缔组织间的连接,导致眼球在胚胎发育时期形成水泡或血泡[15,20].

图1 Fras1蛋白与Frem2蛋白结构
谷氨酸受体相互作用蛋白1(Grip1)是一种胞质支架蛋白,由7个PDZ结构域组成,PDZ结构域与Fras1和Frem2蛋白胞浆区的羧基端作用,从而影响Fras1/Frem蛋白在基底膜的正确定位.Grip1分布在胚胎表皮、眼睑、口腔及鼻腔上皮等组织中,参与跨膜蛋白的转运功能.GRIP1基因丢失可导致细胞与ECM蛋白连接异常[22].
ECM蛋白与细胞间的正常粘连对组织的结构完整和器官发育过程中上皮间质间的相互联系有重要作用[22].三种蛋白均在多种胚胎组织的表皮中高表达,在组织分化过程中既发挥结构功能又与组织发育密切相连[8].Fras1,Frem2和 Grip1蛋白对 Fras1/ Frem蛋白复合物的完整性至关重要,缺失任何一种基因都会导致复合物出现缺陷并引起功能异常.隐眼畸形的出现时间一般在胎儿宫内第4周的眼睑形成过程中[23],胚胎发育过程中上皮-间质间的连接与基因FRAS1,FREM2,GRIP1的生物功能紧密相关.虽然这些蛋白在胚胎发育期的功能是短暂的,会在后期被胶原Ⅶ取代,但均可通过破坏眼睑上皮细胞和下层间充质间的连接而引起患儿眼部异常.因此可以推测隐眼的发生与表皮附着密切相关[20].
3 致病基因突变的研究现状及与隐眼发生的关系
关于隐眼患者的基因突变情况在国内尚无报道,在国外从2006年Slavotinek首次描述了FS患者FRAS1基因的单基因突变[24],到2008年针对33个FS家族的进一步遗传研究发现FRAS1基因11个新突变位点和FREM2基因1个突变位点[25].截止2013年,从FS患者的血液或组织样本中提取DNA进行基因突变检测,共发现FRAS1基因的26种突变.其中大部分突变是移码突变或无义突变、错义突变[12,24,26],但这些检测出的致病基因突变并未证实100%与隐眼症状发生相关.
现总结2017年以前国外文献[12,17,25,27-30]中报道的FS患者致病基因突变情况(基因提取自患者血液或组织样本,测序方法主要包括致病基因直接测序、微阵列比较基因组杂交等,表1).
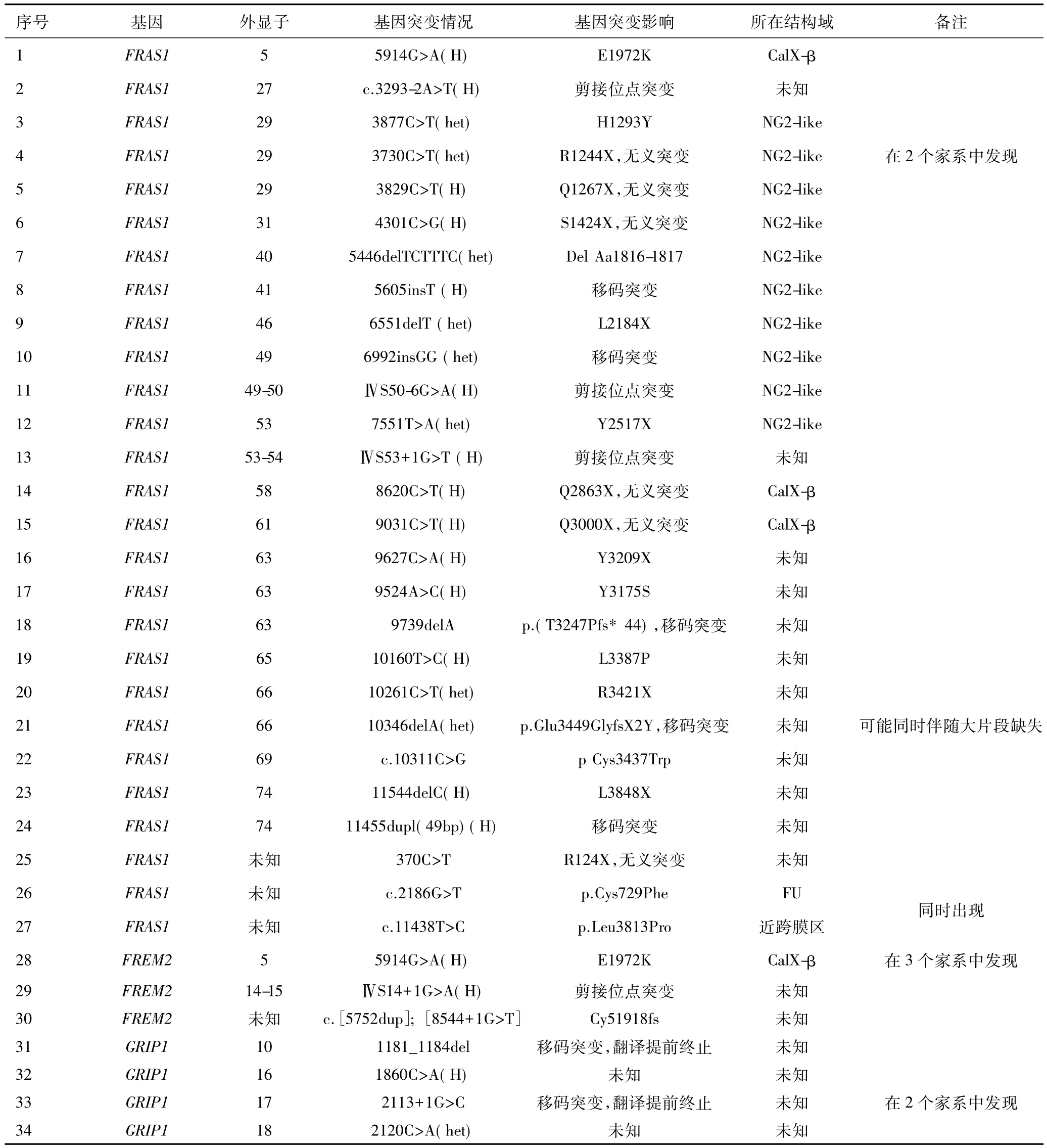
表1 2017年前国外文献报导FS患者致病基因突变情况概览
2008年van Haelst等[25]曾对48位FS患者进行遗传分析,其中来自18个血缘家族的患者遗传连锁分析表明60%的病例与基因FRAS1、FREM2存在关联.后来有研究发现人GRIP1基因发生突变同样可引起经典FS表型,且与FRAS1和FREM2致病机制类似,GRIP1基因突变能够引起胶原Ⅴ和胶原Ⅵ等浓度异常而导致表皮层分离[30].尚未发现不同突变位点导致的蛋白异常结构与FS患者的不同表型有明确关联[11].FS患者中家族性隐眼畸形多见,尚未发现致病基因与种族、地区有明确关联,虽然超过50%的患者存在上述致病基因的突变,但仍有一部分患者的遗传缺陷尚不清楚[12].其中FS临床表现的多样性可能与该综合征的遗传异质性相关,一些合并畸形(如新月形的子宫、肛门闭锁或肛门狭窄等)也可以在没有隐眼表现的其他综合征中出现,暗示修饰基因或三等位基因遗传有可能解释FS中的一些表型变异.
4 遗传致病基因的临床转化应用前景
先天性隐眼等罕见病的受关注度是社会医疗水平进步的重要标尺,而罕见病蕴含的科学价值更是很多常见疾病不可比拟的[31].随着基因测序技术的飞跃发展和广泛普及,以往使医疗工作者陷入困境的各种罕见病正在逐渐被发掘成为一个个的科学宝藏.相比FS综合征,单纯表现为先天性隐眼畸形的病例更为稀少,国内现共有8例报道,国外有40余例,但该罕见病发病的分子机制尚不明确[7,32].单纯性隐眼表型单一,不受其他表型发病机制的干扰,更容易明确致病基因和追踪发病过程,可能对阐明隐眼遗传机制和推动临床转化应用有更为重要的意义.且单纯性隐眼患儿无其他器官系统明显异常,胎儿存活比例更高,明确其发病的分子机制对于隐眼的精准治疗及提高这部分患儿的生活质量具有重大意义.新测序技术和遗传学分析工具的出现将进一步帮助我们揭开隐眼发病机制中的谜团,这也是我们正在努力的研究方向.继续深入了解先天性隐眼的遗传机制,不仅有助于明确先天性隐眼的病因和发病机制,而且可转化应用于孕前咨询(指导隐眼患儿家庭)、孕前筛查、产前诊断以及基因治疗,以避免再次出生无法弥补的先天性缺陷的患儿,对最终推动精准医学的快速发展具有重要的科学价值和社会意义.
[1] Celebi AR,Sasani H.Differentiation of true anophthalmia from clinical anophthalmia using neuroradiological imaging[J].World J Radiol,2014,6(7):515-518.
[2]Zehender W.Eine Missgeburt mit hautuberwachsenen augen oder kryptophthalmus[J].Klin Monatsbl Augenheilkd(in German), 1872,10:225-249.
[3]王 越,李冬梅,陈 涛.先天性隐眼二例[J].中华眼科杂志,2006,42(10):934-935.
[4]Verma AS,Fitzpatrick DR.Anophthalmia and microphthalmia[J].Orphanet J Rare Dis,2007,2:47.
[5]Subramanian N,Iyer G,Srinivasan B.Cryptophthalmos:reconstructive techniques——expanded classification of congenital symblepharon variant[J].Ophthal Plast Reconstr Surg,2013,29(4):243-248.
[6]金丽英,杨东光.双眼隐眼畸形一例[J].中国斜视与小儿眼科杂志,2005,13(4):插页2-3.
[7]Egier D,Orton R,Allen L,et al.Bilateral complete isolated cryptophthalmos:a case report[J].Ophthalmic Genet,2005,26(4): 185-189.
[8]Smyth I,Scambler P.The genetics of Fraser syndrome and the blebs mouse mutants[J].Hum Mol Genet,2005,14 Spec No.2: R269-R274.
[9]De Bernardo G,Giordano M,Di ToroA,et al.Prenatal diagnosis of Fraser syndrome:a matter of life or death?[J].Ital J Pediatr,2015,41:86.
[10]Eskander BS,Shehata BM.Fraser syndrome:a new case report with review of the literature[J].Fetal Pediatr Pathol,2008,27(2): 99-104.
[11]McGregor L,Makela V,Darling SM,et al.Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein[J].Nat Genet,2003,34(2):203-208.
[12]Hoefele J,Wilhelm C,Schiesser M,et al.Expanding the mutation spectrum for Fraser syndrome:identification of a novel heterozygous deletion in FRAS1[J].Gene,2013,520(2):194-197.
[13]Vrontou S,Petrou P,Meyer BI,et al.Fras1 deficiency results in cryptophthalmos,renal agenesis and blebbed phenotype in mice[J].Nat Genet,2003,34(2):209-214.
[14]Pavlakis E,Chiotaki R,Chalepakis G.The role of Fras1/Frem proteins in the structure and function of basement membrane[J].Int J Biochem Cell Biol,2011,43(4):487-495.
[15]Mocan MC,Ozgen B,Irkec M.Bilateral orbito-palpebral cysts in a case of cryptophthalmos associated with Fraser syndrome[J].J AAPOS,2008,12(2):210-211.
[16]Jadeja S,Smyth I,Pitera JE,et al.Identification of a new gene mutated in Fraser syndrome and mouse myelencephalic blebs[J].Nat Genet,2005,37(5):520-525.
[17]Vogel MJ,van Zon P,Brueton L,et al.Mutations in GRIP1 cause Fraser syndrome[J].J Med Genet,2012,49(5):303-306.
[18]Carney TJ,Feitosa NM,Sonntag C,et al.Genetic analysis of fin development in zebrafish identifies furin and hemicentin1 as potential novel fraser syndrome disease genes[J].PLoS Genet,2010,6(4): e1000907.
[19]Richardson RJ,Gebauer JM,Zhang JL,et al.AMACO is a component of the basement membrane-associated Fraser complex[J].J Invest Dermatol,2014,134(5):1313-1322.
[20]Kiyozumi D,Sugimoto N,Sekiguchi K.Breakdown of the reciprocal stabilization of QBRICK/Frem1,Fras1,and Frem2 at the basement membrane provokes Fraser syndrome-like defects[J].Proc Natl Acad Sci U S A,2006,103(32):11981-11986.
[21]Fuchs E,Raghavan S.Getting under the skin of epidermal morpho-genesis[J].Nat Rev Genet,2002,3(3):199-209.
[22]Takamiya K,Kostourou V,Adams S,et al.A direct functional link between the multi-PDZ domain protein GRIP1 and the Fraser syndrome protein Fras1[J].Nat Genet,2004,36(2):172-177.
[23]Amrith S,Lee Y,Lee J,et al.Congenital orbito-palpebral cyst in a case of Fraser syndrome[J].Orbit,2003,22(4):279-283.
[24]Slavotinek A,Li C,Sherr EH,et al.Mutation analysis of the FRAS1 gene demonstrates new mutations in a propositus with Fraser syndrome[J].Am J Med Genet A,2006,140(18):1909-1914.
[25]van Haelst MM,Maiburg M,Baujat G,et al.Molecular study of 33 families with Fraser syndrome new data and mutation review[J].Am J Med Genet A,2008,146A(17):2252-2257.
[26]Nayak SS,Salian S,Shukla A,et al.Variable presentation of Fraser syndrome in two fetuses and a novel mutation in FRAS1[J].Congenit Anom(Kyoto),2017,57(3):83-85.
[27]Ng WY,Pasutto F,Bardakjian TM,et al.A puzzle over several decades:eye anomalies with FRAS1 and STRA6 mutations in the same family[J].Clin Genet,2013,83(2):162-168.
[28]Ozemri Sag S,Gorukmez O,Gorukmez O,et al.A novel mutation in the FRAS1 gene in a patient with Fraser syndrome[J].Genet Couns,2015,26(1):21-27.
[29]Ogur G,Zenker M,Tosun M,et al.Clinical and molecular studies in two families with Fraser syndrome:a new FRAS1 gene mutation,prenatal ultrasound findings and implications for genetic counselling[J].Genet Couns,2011,22(3):233-244.
[30]Schanze D,Kayserili H,Satkin BN,et al.Fraser syndrome due to mutations in GRIP1——clinical phenotype in two families and expansion of the mutation spectrum[J].Am J Med Genet A,2014,164A(3):837-840.
[31]Schieppati A,Henter JI,Daina E,et al.Why rare diseases are an important medical and social issue[J].Lancet,2008,371(9629): 2039-2041.
[32]Slavotinek AM,Tifft CJ.Fraser syndrome and cryptophthalmos: review of the diagnostic criteria and evidence for phenotypic modules in complex malformation syndromes[J].J Med Genet,2002,39(9): 623-633.
R770.4
A
2095-6894(2017)08-01-05
2017-02-14;接受日期:2017-03-03
国家自然科学基金面上项目(81770967);国家自然科学基金重大研究计划培育项目(91546101);广东省自然科学杰出青年基金项目(2014A030306030);广东省高等学校优秀青年教师培养计划 (YQ2015006);广东省高层次人才特殊支持计划(2014TQ01R573)
张夏茵.硕士生.E-mail:643135449@qq.com
林浩添.博士,研究员,副教授,副主任医师.研究方向:眼科学.E-mail:haot.lin@hotmail.com
猜你喜欢
河北果树(2021年4期)2021-12-02
天津医科大学学报(2021年4期)2021-08-21
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年2期)2021-01-18
心肺血管病杂志(2020年5期)2021-01-14
医药前沿(2020年20期)2020-11-10
河北农业科学(2019年6期)2019-03-21
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
医学研究杂志(2015年12期)2015-06-10
