
深圳地区小儿α和β地中海贫血基因类型分析*
2017-09-11 09:39:10任振敏蔡德丰肖伟伟徐刚刘永秋马东礼
临床检验杂志 2017年8期
任振敏,蔡德丰,肖伟伟,徐刚,刘永秋,马东礼
(深圳市儿童医院检验科, 广东深圳 518038)
·临床实验研究·
深圳地区小儿α和β地中海贫血基因类型分析*
任振敏,蔡德丰,肖伟伟,徐刚,刘永秋,马东礼
(深圳市儿童医院检验科, 广东深圳 518038)
目的 分析深圳地区小儿地中海贫血的基因类型及突变频率,为该疾病基因诊断及遗传咨询提供参考。方法 回顾性分析1 206例疑似地中海贫血患儿,采用跨越断裂点PCR(Gap-PCR)检测缺失型α地中海贫血,反向点杂交(RDB)技术检测α和β地中海贫血基因点突变,巢式PCR检测疑似HKαα的样本并用基因测序验证疑似罕见地中海贫血的样本。结果 1 206例病例中共检出927例地中海贫血(76.9%),其中489例(40.5%)α地中海贫血,主要以--SEA/αα为主(与75.1%);406例(33.7%)β地中海贫血,主要以IVS-2-654杂合子和CD41-42杂合子为主(分别占35%和32.5%)。检出αβ复合型地中海贫血32例(2.7%)。此外,发现HKαα/ααQS、α-地中海贫血突变基因类型CD61(AAG→TAG)/--SEA、β地中海贫血基因突变类型CD5(CCT→C)各1例。结论 深圳地区地中海贫血患儿基因突变类型复杂多样,且存在多例罕见病例。
α-地中海贫血;β-地中海贫血;基因型
地中海贫血(以下简称“地贫”)是一组极易导致致死、致残的遗传性血液病。近年来,为了防止重型地贫患儿的出生,深圳地区对于育龄人群的地贫进行大量的筛查研究[1-2],但对患儿缺乏较为系统的地贫基因分析报道。因此,本研究旨在阐明深圳地区小儿的地贫基因携带情况,以期能够更好地指导本地区地贫防控,并为该病的资料累积提供数据。
1 材料和方法
1.1 研究对象 收集2016年3月至2017年2月于深圳市儿童医院就诊的门诊、住院以及体检的疑似地贫的初诊患儿共1 206例,男739例,女467例,年龄(2.9±2.7)岁(7 d~14岁)。诊断根据《血液病诊断及疗效标准》中“珠蛋白生成障碍性贫血诊断标准”以及《地中海贫血预防控制操作指南》中“α和β地中海贫血的临床分类和临床特征”[3-4]。纳入标准:(1)患儿实验室血液学指标异常(平均红细胞体积MCV<80 fL和/或平均红细胞血红蛋白含量MCH<27 pg);(2)Hb电泳提示有地贫(HbA2≤2.5%,提示α地贫可疑,可出现H带或Bart′s带;HbA2≥3.5%,提示β地贫可疑,可出现E带);(3)家族中有地贫的患儿、地贫基因携带者。排除标准:已经输血治疗的地贫患儿。本研究经我院医学伦理学委员会批准,患儿的监护人知情同意。
1.2 主要的仪器与试剂 XS-1000i全自动五分类血球计数仪(日本Sysmex公司),Capillarys2全自动毛细管电泳分析仪(法国Sebia公司),PCR扩增仪、电泳仪和凝胶成像系统(美国Bio-Rad公司),5430
小型台式高速离心机(德国Eppendorf公司),NanoDrop 2000c微量分光光度计(美国Thermo公司),YN-H16恒温杂交仪(深圳亚能公司);全血DNA快速提取试剂盒和珠蛋白生成障碍性贫血基因检测试剂盒(深圳亚能公司)。
1.3 标本采集 采集疑似地贫患儿入院或门诊时静脉血2 mL,体检患儿于体检时采集静脉血2 mL,EDTA-K2抗凝,标本置-80 ℃保存。
1.4 地贫的实验室筛查 用XS-1000i全自动五分类血球计数仪及配套试剂检测红细胞计数(RBC)、血红蛋白(Hb)、平均红细胞体积(MCV)、平均红细胞血红蛋白量(MCH)。Capillarys2全自动毛细管电泳分析仪及配套试剂检测血红蛋白A(HbA)、血红蛋白A2(HbA2)、胎儿血红蛋白(HbF)以及异常Hb。均按仪器及试剂盒说明书操作及结果判读。
1.5 地贫基因检测
1.5.1 基因组DNA提取 取200 μL的静脉血标本,按照全血DNA快速提取试剂盒说明书操作提取基因组DNA。Nanodrop2000c微量分光光度计检测DNA浓度和纯度,取浓度为50~100 ng/μL、吸光度(A260/280 nm)为1.7~1.9的样本用于后续实验。
1.5.2 Gap-PCR检测缺失型α-地贫 缺失型α-地贫检测试剂盒说明书操作检测--SEA缺失、-α3.7缺失和-α4.2缺失 3种基因缺失。PCR总反应体积为25 μL,包括PCR反应液21 μL,DNA模板4 μL。PCR循环参数:96 ℃ 5 min;98 ℃ 45 s,65 ℃ 90 s,72 ℃ 3 min,10个循环;98 ℃ 30 s,65 ℃ 45 s,72 ℃ 3 min,25个循环;72 ℃ 10 min;4 ℃保存。PCR产物经10 g/L琼脂糖凝胶电泳,根据电泳条带大小判断基因型。以扩增出2 000、1 700、1 400、1 200、900 bp条带分别对应于右侧缺失(-α3.7)、无缺失、左侧缺失(-α4.2)、东南亚缺失(--SEA)、泰国型缺失(--THAI)。
1.5.3 RDB-PCR检测α和β地贫非缺失型点突变 按照α和β地贫非缺失型点突变检测试剂盒说明书操作检测3种α2珠蛋白基因突变类型[αWS(CD122),CAC-CAG;αQS(CD125),CTG-CCG;αCS(CD142),TAA-CAA]和17种β珠蛋白基因突变类型[-28、-29、-30、-32、 CD14-15、CD17、CD26(βE)、CD27-28、CD31、 CD41-42、CD43、CD71-72、IVS-Ⅰ-1、IVS-Ⅰ-5、IVS-Ⅱ-654、CAP+1、起始密码子(initiation condon)]。PCR总反应体积为25 μL,包括PCR反应液23 μL,DNA模板2 μL。循环参数:50 ℃ 15 min,95 ℃ 10 min;94 ℃ 1 min,55 ℃ 30 s,72 ℃ 30 s,35个循环;72 ℃ 5 min。PCR产物通过杂交膜条斑点显色确定基因型。结果判读:野生斑点全部显色且无异常突变斑点同时显色为野生基因型;突变斑点及其相应野生斑点同时显色为突变杂合子。β地贫突变:斑点显色而相应野生斑点不显色为突变纯合子;α地贫突变:若α地贫基因检测缺失,则为α点突变类型/α缺失型类型;若α地贫基因检测未发现缺失,则为突变纯合子。
1.5.4 罕见型地贫基因检测 将试剂盒未检出且怀疑HKαα及罕见地贫基因的样本送至深圳亚能公司进行巢式PCR和基因测序检测。测序结果与野生型序列进行比对分析,确定有无碱基突变。
1.6 统计学分析 用Excel 2007软件进行数据录入、整理,采用描述性统计分析深圳地区小儿α和β地贫基因突变类型及频率分布。
2 结果
2.1 地贫基因检测结果 在1 206例疑似地贫的病例中,确诊为地贫患儿927例(76.9%)。其中α地贫489例(40.5%);β地贫406例(33.7%);αβ复合型地贫32例(2.7%)。
2.2 地贫基因类型的分布特征和突变频率 489例α地贫中共检出17种α地贫突变基因类型,以--SEA/αα为主(75.1%)。406例β地贫中共检出24种突变基因类型,以IVS-2-654(C→T)杂合子和CD41-42(-TCTT)杂合子为主(分别占35%和32.5%)。32例αβ复合型地贫以IVS-2-654/N+--SEA/αα和IVS-2-654/N+-α3.7/αα为主,各占15.6%。地贫基因突变类型和构成比见表1,突变频率见表2。
2.3 罕见地贫基因的检测结果 巢氏PCR检出1例罕见的HKαα/ααQS,见图1。基因测序检出α-地中海贫血突变基因类型CD61(AAG→TAG)/--SEA、β地中海贫血基因突变类型CD5(CCT→C)各1例,见图2。3例患儿的实验室检测结果见表3。

表1 地中海贫血基因类型与构成比

续表
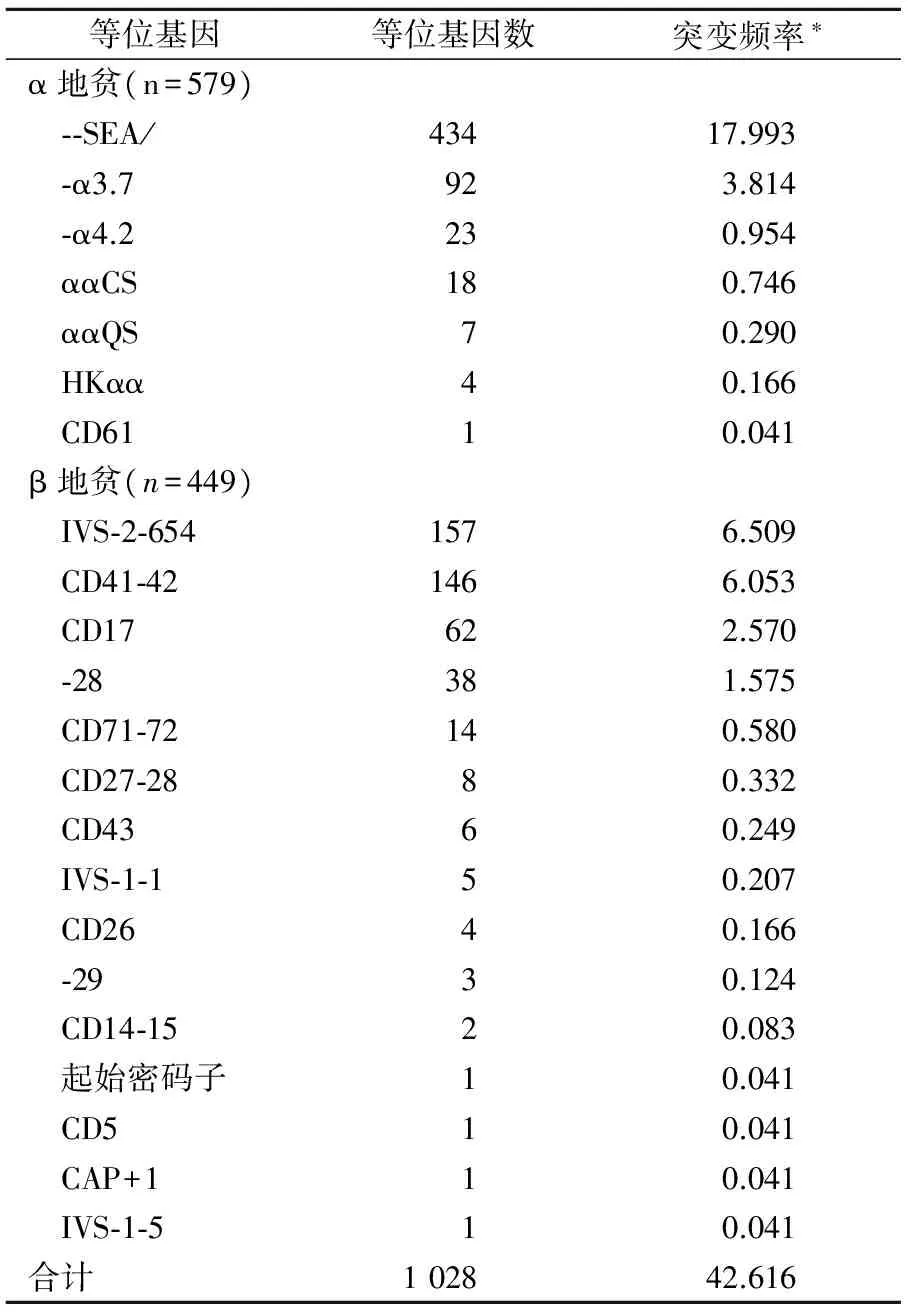
表2 α和β地中海贫血基因突变频率
注:*,突变频率=等位基因数/被研究的染色体总数(1 206×2)。
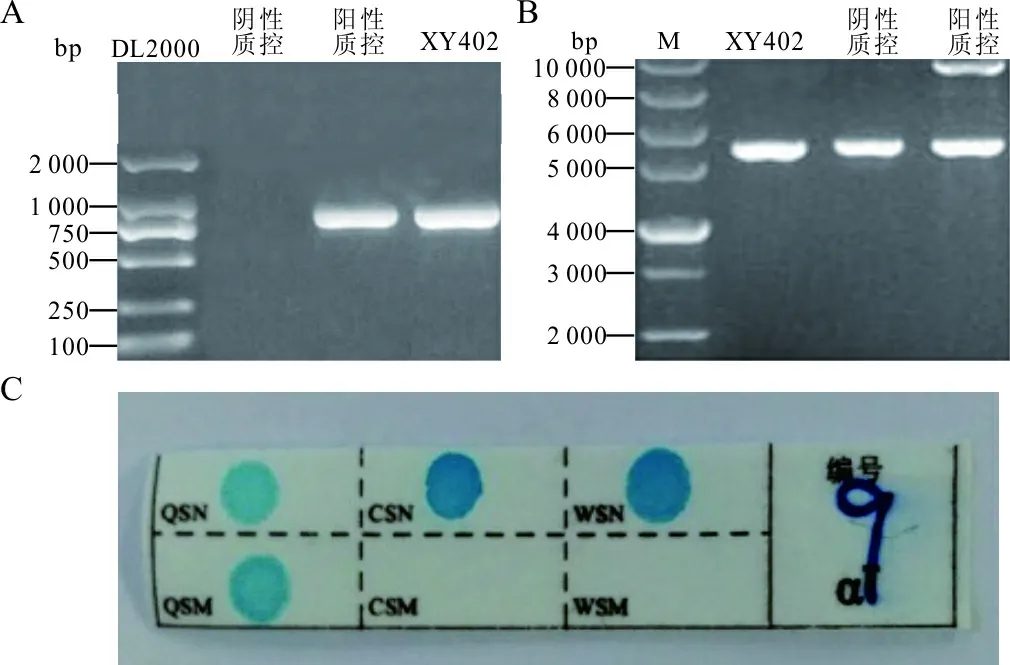
注:A,第1轮GAP-PCR电泳结果,1~3泳道分别为阴性质控(DL 2000、HKαα或αααanti4.2)、阳性质控(HKαα或αααanti4.2);B,第2轮GAP-PCR电泳结果,1~3泳道分别为DNA marker,阴性质控(HKαα)、阳性质控(αααanti4.2); C,α突变结果为QS杂合。
图1 罕见地贫基因类型的巢氏PCR检测结果
3 讨论
本研究共检出17种α地贫和24种β地贫突变基因类型,虽然以轻型--SEA/αα、IVS-2-654(C→T)杂合、CD41-42(-TCTT)杂合为主,但是中间型、重型和复合型地贫也有检出。HbH病(中间型)是出生患儿中最严重的α地贫类型,尤其是HbH-CS或QS多伴有较重的贫血症状[5]。本研究检出HbH病55例,阳性率达11.2%,可能是由于HbH病常导致中、重度贫血,易被家长发现而就诊有关。β地贫纯合子和双重杂合子一般呈中间型/重型地贫。本研究共检出纯合子11例,双重杂合子4例,提示应加强本地区育龄人群β地贫的筛查,并对高危人群进行基因诊断,以避免该类患儿的出生。αβ复合型地贫血液学表现为β地贫的特点[6],因此对已确诊的β-地贫患儿要进行α地贫基因检测,以免漏诊。

注:A,CD61由野生型的AAG突变为TAG;B,β基因CD5由野生型CCT突变为C,即框内标记(-CT)。
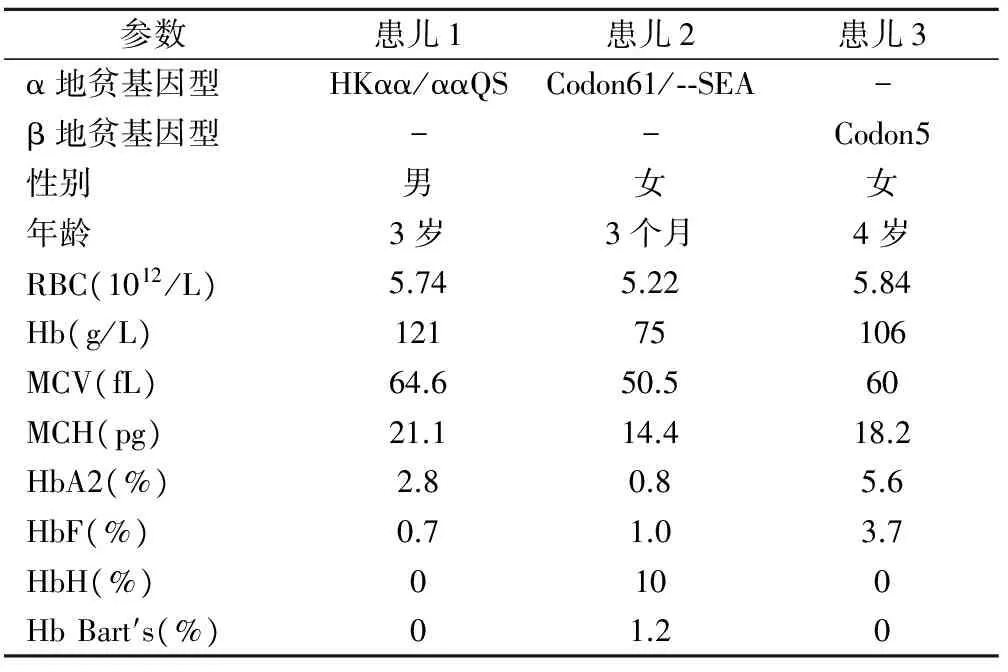
图2 罕见地贫基因类型的测序检测结果
除了常见的基因类型外,本研究还发现了HKαα地贫。HKαα于2005年被首次发现[7],此基因型是在1条染色体上同时含有-α3.7和αααanti4.2融合基因片段,常规地贫基因检测结果为-α3.7。刘朔婕等[8]通过巢式PCR检测发现闽南地区HKαα在地贫疑似患者中的检出率为0.11%, 与广西、广东地区该基因的检出率类似[9],印证了Shang等[10]关于HKαα并非罕见现象的推测。因此,为避免HKαα基因型误诊为-α3.7,要求检验人员遇到常规地贫基因检测为-α3.7的样本时,一定要结合表型与家系资料做好HKαα基因的检测工作,避免漏诊和误诊。本研究中发生的HKαα/ααQS在国内仅见姚亚超等[11]在2015年首次报道。此病例利用常规方法检测时扩增出-α3.7条带,如果是-α3.7缺失,那么在α突变的检测中,QS的突变应该是纯合子,与结果为杂合子不符,这种情况下要考虑存在HKαα基因。由于这种基因融合血液学表型比缺失型较轻,此病例的血液学结果显示除了小细胞外其他均在参考范围内,与陈文璟等[12]报道的HKαα /--SEA的临床表象类似--SEA /αα相符。
此外,本研究在α地贫患儿中还发现了1例尚未见报道的CD61与--SEA双重突变,此病例的常规基因检测时只发现了--SEA/αα,而血常规结果表现为中度贫血,并且Hb电泳结果示存在HbH带和Hb Bart′s带,临床提示为HbH病,所以考虑除了--SEA外,还存在其他的突变点。同时,在β地贫患儿中也发现了1例罕见的病例,其表现为轻度贫血,毛细管电泳证实其HbA2水平符合β地贫的特点,但常规检测方法未发现基因突变,经测序证实为CD5β0杂合突变。
综上所述,深圳地区的地贫基因型复杂多样,可能与流动人口增多有关,但主要人群来自广东省,故常见的地贫基因类型与广东省的其他地区基本一致[13-15]。罕见地贫的检出提示利用常规方法诊断地贫时,一定要结合血液学表型综合分析,一旦遇到不相符的病例,要高度怀疑携带罕见地贫基因类型,重视罕见基因突变类型的检测。
感谢深圳市亚能生物科技有限公司所提供的技术支持。
[1]文艳, 刘爱胜, 阳建. 深圳地区未婚青年人群地中海贫血基因携带率现状调查[J]. 中国优生与遗传杂志, 2016,24(6):13-14.
[2]袁晖, 吴维青, 吴晓霞,等. 深圳地区育龄人群地中海贫血基因型分布调查[J]. 中山大学学报(医学科学版), 2012, 33(4):553-557.
[3]张之南,沈悌.血液病诊断及疗效标准[M].第3版.北京:科学出版社,1999.
[4]徐湘民.地中海贫血预防控制操作指南[M].北京:人民军医出版社,2011.
[5]孙曼娜, 熊符, 张新华,等. 血红蛋白Constant Spring的表型与基因型分析[J]. 中华医学遗传学杂志, 2010, 27(5):481-483.
[6]Wee YC,Tan KL,Kuldip K,etal.Alpha-thalassaemia in assoaiation with beta-thalassaemia patients in Malaysia:a study on the co-inheritance of both disorders[J].Community Genet, 2008,11(3):129-134.
[7]Wang W, Chan AY, Chan LC,etal. Unusual rearrangement of the alpha-globin gene cluster containing both the -alpha3.7 and alphaalphaalphaanti-4.2 crossover junctions: clinical diagnostic implications and possible mechanisms.[J]. Clin Chem, 2005, 51(11):2167-2170.
[8]刘朔婕, 孙鸣, 黄宇,等. 闽南地区罕见α地中海贫血基因型分析[J]. 医学分子生物学杂志, 2016, 13(6):311-316.
[9]阙婷, 李东明, 李旺,等. 香港型α-地中海贫血杂合子地中海贫血经验分析和分子机制[J]. 中国优生与遗传杂志, 2014,26(12):19-20.
[10]Shang X, Li Q, Cai R,etal. Molecular characterization and clinical presentation of HKαα and anti-HKαα alleles in southern Chinese subjects[J]. Clini Genet, 2013, 83(5):472-476.
[11]姚亚超, 李磊, 李泽泳,等. HK型α珠蛋白生成障碍基因诊断及其家系分析[J]. 检验医学与临床, 2015,12(12):1672-1675.
[12]陈文璟, 温旺荣, 苏运钦,等. 用巢氏PCR检测防止香港型地中海贫血漏诊[J]. 临床检验杂志, 2014, 32(9):713-715.
[13]刘玲, 蒋玮莹, 许世艳,等. 广东地区地中海贫血致病基因的基因型及β珠蛋白基因多态性研究[J]. 中华血液学杂志, 2013, 34(7):595-599.
[14]黄秀霞, 梁志江, 刘志祥,等. 广东省河源市789户核心家庭α-和β-地中海贫血基因突变研究[J]. 中国生育健康杂志, 2014, 25(3):281.
[15]唐玉芬, 谭满胜, 聂俊玮. 广东省茂名地区地中海贫血基因型分析[J]. 分子诊断与治疗杂志, 2014, 6(3):187-190.
(本文编辑:许晓蒙)
Genotype analysis of α-thalassemia and β-thalassemia in child patients of Shenzhen region
RENZhen-min,CAIDe-feng,XIAOWei-wei,XUGang,LIUYong-qiu,MADong-li
(DepartmentofClinicalLaboratory,ShenzhenChildren'sHospital,Shenzhen518038,Guangdong,China)
Objective To investigate the genotype and mutation frequency of thalassemia in child patients of Shenzhen region so as to provide evidences for the gene diagnosis and genetic counseling of thalassemia. Methods A total of 1 206 child patients suspected with thalassemia were retrospectively analyzed. The gene deletion of α-thalassemia was detected by Gap-PCR. The point mutations of α-thalassemia and β-thalassemia were determined by reverse dot blot(RDB)-PCR. The specimens suspected with HKαα and rare gene mutations were determined with nested PCR and gene sequencing, respectively. Results The detection rate of thalassemia was 76.9%(927/1 206). Among them, α-thalassemia accounted for 40.5%(489/1 206), and --SEA/αα was the most common gene mutation(75.1%); β-thalassemia accounted for 33.7%(406/1 206), and the main IVS-2-654(C→T) and CD41-42(-TCTT) heterozygous mutations accounted for 35% and 32.5%, respectively. In addition, there were 32(2.7%) β-thalassemia patients with α-thalassemia mutation, 1 patient with HKαα/ααQS, 1 α-thalassemia patient with CD61(AAG→TAG)/--SEA and 1 β-thalassemia patient with CD5(CCT→C). Conclusion The are complicated gene mutation types and rare gene mutations of thalassemia in child patients of Shenzhen region.
α-thalassemia; β-thalassemia; genotype
10.13602/j.cnki.jcls.2017.08.12
深圳病原体高通量基因测序技术工程实验室项目(深发改[2014]1712号)。
任振敏,1981年生,女,主管技师,硕士,主要从事遗传病的分子研究。
马东礼,主任技师,E-mail:madl1234@126.com。
R33
A
2017-03-23)
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
科学大众(2021年6期)2022-01-01 00:45:52
基层中医药(2021年4期)2021-07-22 07:15:26
基层中医药(2021年4期)2021-07-22 07:15:18
中国生殖健康(2020年2期)2021-01-18 02:51:26
家庭医学(下半月)(2020年6期)2020-08-24 07:46:14
小学生导刊(2018年13期)2018-06-29 03:49:00
现代检验医学杂志(2015年6期)2015-02-06 01:44:02
实验动物与比较医学(2014年5期)2014-02-28 14:53:10
河南医学研究(2014年5期)2014-02-27 14:52:41
