
替米沙坦对高血压合并阿尔茨海默病患者血清Aβ蛋白、Tau蛋白和炎性细胞因子的影响
2017-08-01 00:53王丹琼卜星彭鹿育萨
山西医科大学学报 2017年6期
王丹琼,卜星彭,王 蕾,霍 晋,鹿育萨
(山西大医院老年医学科,太原 030032;*通讯作者,E-mail:lystysx@126.com)
替米沙坦对高血压合并阿尔茨海默病患者血清Aβ蛋白、Tau蛋白和炎性细胞因子的影响
王丹琼,卜星彭,王 蕾,霍 晋,鹿育萨*
(山西大医院老年医学科,太原 030032;*通讯作者,E-mail:lystysx@126.com)
目的 观察替米沙坦对高血压合并阿尔茨海默病(AD)患者血清Aβ蛋白、Tau蛋白和炎性细胞因子的影响。 方法 收集高血压合并阿尔茨海默病患者62例,分别用替米沙坦联合盐酸多奈哌齐(治疗组,n=31)、左旋氨氯地平联合盐酸多奈哌齐(对照组,n=31)治疗。治疗前、治疗48周后分别对两组患者进行血清Aβ1-42、Tau蛋白、IL-1β、IL-6、TNF-α检测。 结果 治疗48周后,两组患者血清Aβ1-42 、Tau蛋白、IL-1β、IL-6、TNF-α水平均低于治疗前(P<0.05),治疗组血清Aβ1-42蛋白测量值高于对照组,而Tau蛋白和炎性细胞因子IL-1β、IL-6、TNF-α水平治疗组低于对照组(P<0.05)。 结论 替米沙坦可延缓AD患者血清Aβ1-42下降,降低血清中Tau蛋白、IL-1β、IL-6、TNF-α水平。
替米沙坦; 阿尔茨海默病; 高血压; Aβ蛋白; Tau蛋白; 炎性细胞因子
阿尔茨海默病(Alzheimer’s disease,AD)是多发于老年人的神经系统变性疾病,临床上以进行性记忆下降、认知障碍及行为异常为特征,典型的病理学表现为β淀粉样(Aβ)蛋白沉积、神经原纤维缠结(NFTs)、神经元减少及轴突和突触异常等,目前认为Aβ蛋白沉积是AD患者最典型的病理生理改变,其诱导的炎症反应也可能是AD发病的重要机制之一,因此炎症在AD发病机制中的作用逐渐受到人们的重视[1,2]。NFTs是AD的另一主要病理改变,其主要成分是异常磷酸化的微管相关Tau蛋白。近年有研究显示血管紧张素Ⅱ(AngⅡ)通过激活血管紧张素Ⅰ(AT1)受体,引起小鼠脑内不同部位的Tau蛋白发生磷酸化,促进NFTs产生。血管紧张素Ⅱ受体拮抗剂 (ARB) 类降压药物替米沙坦可通过部分激活脑中过氧化物酶体增殖物激活受体(PPARγ)而抑制星型胶质细胞和小胶质细胞介导的炎症反应,增加Aβ的清除,减少磷酸化Tau蛋白产生[3-5]。与钙离子拮抗剂类(CCB)降压药左旋氨氯地平相比,替米沙坦能减缓认知功能的下降[6]。本研究探讨替米沙坦联合盐酸多奈哌齐对高血压合并AD患者治疗前后血清Aβ蛋白、Tau蛋白和多种炎性细胞因子水平变化,为今后AD患者的治疗提供更多依据。
1 资料与方法
1.1 研究对象
收集2012-10~2015-10就诊于山西大医院老年医学科门诊、病房的高血压合并AD患者62例,男38例,女24例;年龄58-81岁,平均(69.2±3.4)岁;病程6-36个月,平均(22.8±3.6)月。入选标准:①认知障碍的首发表现为近记忆力下降;②符合2011年中华医学会《中国痴呆与认知障碍诊治指南》的诊断标准[7];③临床均表现为不同程度认知功能损伤;④筛选时全部病例均进行简易精神状态量表(MMSE)测试,得分12分≤MMSE≤24分;⑤高血压的诊断采用2010年中国高血压防治指南的标准;⑥所有入组患者均签署知情同意书。排除标准:①均经头颅CT或MRI诊断证实除外急性脑血管疾病;②入组前3个月内发生脑血管意外事件。将62例患者按照随机数字表法分为治疗组与对照组。两组患者一般资料比较,差异无统计学意义(P>0.05),具有可比性。
1.2 方法
1.2.1 治疗方法 入选62例患者,根据随机数字表分为治疗组和对照组,每组各31例。治疗组治疗方案为替米沙坦40 mg/次+盐酸多奈哌齐5 mg/次,口服,每日1次,连续服药48周;对照组治疗方案为左旋氨氯地平2.5 mg/次+盐酸多奈哌齐5 mg/次,口服,每日1次,连续服药48周。
1.2.2 标本的收集 所有受试者均在基线期及治疗后48周时抽取血样,受试者至少静坐15 min后,于早餐前(7:30-9:00)抽取清晨空腹肘静脉血5 ml,4 000 r/min即时离心(离心半径=8 cm)10 min,取血清置于-80 ℃低温冰箱内冻存。
1.2.3 标本的测定 采用双抗体夹心酶联免疫吸附试验,检测血清中Tau蛋白和β淀粉样蛋白1-42(Aβ1-42)、IL-1β、IL-6、TNF-α的含量。血清IL-1β、IL-6、TNF-α浓度测定试剂盒为MBI公司产品,Tau蛋白试剂盒由瑞士Bachem公司提供,Aβ1-42试剂盒由Abcam公司提供,均购自深圳晶美生物工程有限公司。
1.3 观察指标
观察治疗前、后两组患者血清Aβ1-42蛋白、Tau蛋白、IL-1β、IL-6、TNFα水平变化。
1.4 统计学方法
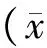
2 结果
2.1 组内治疗前后指标比较
同组内治疗前后比较,治疗组(替米沙坦联合盐酸多奈哌齐治疗组)在治疗48周后,血清Aβ1-42蛋白、Tau蛋白和炎性细胞因子水平下降,差异有统计学意义(P<0.01,见表1)。对照组(左旋氨氯地平联合盐酸多奈哌齐组)在治疗48周后,血清Aβ1-42蛋白、Tau蛋白和炎性细胞因子水平下降,差异有统计学意义(P<0.05,见表1)。
2.2 组间治疗后指标比较
治疗后组间比较,治疗组在治疗48周后血清Aβ1-42蛋白检测值高于对照组,Tau蛋白和炎性细胞因子水平显著低于对照组,差异有统计学意义(P<0.05,见表1)。

组别时间Aβ1⁃42Tau蛋白IL⁃1βIL⁃6TNF⁃α治疗组治疗前1294±1852287±273349±5264±081477±166治疗后1145±167∗∗1911±278∗∗270±52∗∗ 45±08∗∗1075±168∗∗对照组治疗前1291±1572307±298353±5366±091478±155治疗后981±183∗#2123±304∗#314±53∗# 56±09∗#1282±149∗#
与同组治疗前比较,*P<0.05,**P<0.01;治疗后与对照组比较,#P<0.05
2.3 不良反应
本研究治疗期间未出现明显不良反应,无退出研究者。
3 讨论
随着人口社会老龄化的逐步加剧,高血压病和AD的发病率逐年升高,二者并存的患病率也有上升趋势。临床常用的ARB类降压药物替米沙坦,不仅具有拮抗AT1受体的作用,同时也能部分激活PPARγ,其双靶点的活性决定了其不仅具有良好的降压作用,而且也具有其他靶器官保护的特性。替米沙坦发挥神经保护作用与其激活PPARγ进而抑制或改善凋亡、氧化应激以及神经炎症密切相关[8],其可能机制是通过激活PPARγ从而抑制小胶质细胞炎症反应而发挥神经保护作用[9-12]。因此,我们选用替米沙坦观察其对高血压合并AD的治疗作用。
目前认为Aβ沉积及其诱导的炎症反应是AD发病的重要机制之一。研究显示,AD患者的脑组织、脑脊液和血液中发现多种炎性因子如IL-1β、IL-6、TNF-α等的变化[1]。而异常磷酸化的Tau蛋白是NFTs形成的基础。由于脑脊液标本特异性高,国内外相关研究比较多,而血清的相关指标研究的较少,虽然血液标本的特异性不如脑脊液,但血液标本的采集具有方便快捷、易于接受、安全性高等优点,是临床来源最广泛的样本。在本研究中,我们主要观察了替米沙坦对AD患者治疗后血清Aβ1-42蛋白、Tau蛋白和炎性细胞因子IL-1β、IL-6、TNFα水平,并与对照组进行比较。结果发现,治疗组血清Aβ1-42测量值高于对照组,而血清Tau蛋白、IL-1β、IL-6、TNF-α均较对照组明显下降,差异有明显统计学意义。结果提示,替米沙坦可能通过减少Aβ沉积、抑制炎症反应、减轻脑内Tau蛋白的磷酸化起作用,且较对照组疗效显著。
AD的两大组织病理学特点为以Aβ沉积为核心的老年斑和高度磷酸化Tau蛋白聚集形成的神经元纤维缠结[13]。Aβ1-40和Aβ1-42是Aβ肽在体内的两个主要亚型,其中,Aβ42 的神经毒性最强,容易聚集形成淀粉样蛋白,且可以促使老年斑形成增多,与AD 的关系最为密切[14]。颅内Aβ1-42 通过高级糖化终产物受体(receptor for advanced glycation end products,RAGE) 及低密度脂蛋白受体相关蛋白(low-density lipoprotein receptor-related protein 1,LRP-1)介导的跨膜转运以通过血脑屏障[15]。胰岛素降解酶(insulin-degrading enzyme,IDE) 在Aβ1-42降解中起到重要作用,研究证实IDE功能缺陷可导致脑中Aβ降解障碍,而IDE过度表达可降低Aβ水平、延迟或完全预防脑中淀粉样斑块的形成[16]。IDE表达可能受PPARγ调控[17]。激活PPARγ可使IDE转录增加,从而增加的Aβ1-42降解。本研究结果显示,两组患者治疗后血清Aβ1-42水平均有下降,但替米沙坦治疗组患者血清Aβ1-42测量值高,差异有统计学意义,其原因可能是随着病理进程脑中致密的淀粉样沉积增多,故其血液浓度相应降低[18]。
Tau蛋白属于大脑磷酸化蛋白,其具有维持微管装配过程稳定、连接轴突微管的作用,同时与细胞内物质转运有密切关系。当脑组织受损时,Tau蛋白可被磷酸化并丧失维持微管运输的功能,并形成NFTs沉积在脑血管中,导致患者出现认知功能障碍[16]。有研究显示AngⅡ通过激活AT1R,引起小鼠脑内不同部位的Tau蛋白发生磷酸化,促进NFTs产生。在本研究中,与对照组相比,替米沙坦治疗组血清Tau蛋白水平显著下降,其差异有统计学意义。
近年来,AD 发病的炎症机制也越来越被大家所重视,有人提出慢性炎症反应可能是其另一重要的病理特征[19,20]。在炎性细胞因子网络中,促炎和抗炎细胞因子的平衡是机体产生适度免疫应答的关键,IL-1β、IL-6、TNF-α被认为是典型的促炎细胞因子。国外研究表明,促炎介质的释放会促进海马神经元凋亡、抑制长时程增强(LTP),导致动物出现认知障碍,IL-1β、IL-6、TNF-α等促炎介质通过直接或间接途径影响中枢神经功能,IL-1β和TNF-α可通过脑室区域的血脑屏障进入大脑内,引起中枢神经炎性反应[21]。研究发现炎症介质TNF-α刺激可以引起PPARγ下降;另外,已证实PPARγ配体具有抗炎效应,激活PPARγ可减轻炎症反应,使炎症因子表达减少。研究还表明随着IL-1、TNF-α等表达上调,PPARγ表达是下降的,这提示炎症因子可能下调PPARγ表达[17,18]。本研究发现,与对照组相比,替米沙坦治疗组血清炎性因子IL-1β、IL-6、TNF-α均有下降,且差异有统计学意义。这些结果提示AD 患者体内可能存在不同程度的炎症反应,而替米沙坦可能通过激活PPARγ,抑制炎性因子的表达,减轻中枢神经炎性反应。
综上所述,本研究结果认为替米沙坦可以减缓Aβ1-42的沉积,增加Aβ1-42的清除,抑制炎症反应;可能通过拮抗AT1受体,减轻Tau蛋白的磷酸化,使NFTs生成减少,从而延缓AD的病程进展,改善认知。
[1] Ghosh S,Wu MD,Shafiel SS,etal.Sustained interleukin-1 p over expression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model[J].J Neurosci,2013,33(1 1):5053-5064.
[2] 解建波,孙志清.淀粉样蛋白、炎性细胞因子与阿尔茨海默病[J].国外医学(神经病学神经外科学分册),2002,29(6):503-505.
[3] Suzuki Y,Ruiz-Ortega M,Lorenzo O,etal.Inflammation and angiotensin II[J].Biochem Cell Biol,2003,35: 881-900.
[4] Camacho IE,Serneels L,Spittaels K,etal.Peroxisome-proliferator-activated receptor gamma induces a clearance mechanism for the amyloid-beta peptide[J].Neuroscience,2005,24: 10908-10917.
[5] Gurley C,Nichols J,Liu S,etal.Microglia and astrocyte activation by toll-like receptor ligands: modulation by PPAR-gamma agonists[J].PPAR Res,2008,45: 31-40.
[6] Arun KH,Kaul CL,Ramarao P,etal.High glucose concentration augments angiotensin II mediated contraction via AT1 receptors in rat thoracic aorta[J].Pharmacol Res,2004,50(6): 561-568.
[7] 贾建平,王荫华,魏翠柏,等.中国痴呆与认知障碍诊治指南[J].中华医学杂志,2011,91(14):940-945.
[8] Kasahara Y,Taguchi A,Uno H,etal.Telmisartan suppresses cerebral injury in a murine model of transient focal ischemia[J].Brain Res,2010,1340: 70-80.
[9] Aloisi F.Immune function of microglia[J].Glia,2001,36(2): 165-179.
[10] Pearson G,Robinson F,Beers GT,etal.Mitogen-activated protein (MAP) kinase pathways: regulation andphysiological functions [J].Endocr Rev,2001,22(2): 153-183.
[11] Chow YL,Lee KH,Vidyadaran S,etal.Cardamonin from Alpinia rafflesiana inhibits inflammatory in IFN-gamma/LPS-stimulated BV2 microglia via NF-kappaB signaling pathway[J].Int Immunopharmacol,2012,12(4): 657-65.
[12] Kanarek N,Ben-Neriah Y.Regulation of NF-κB by ubiquitination and degradation of the IκBs[J].Immunol Rev,2012,246(1): 77-94.
[13] Meraz-Ríos MA,Toral-Rios D,Franco-Bocanegra D,etal.Inflammatory process in Alzheimer's Disease[J].Front Integr Neurosci,2013,7(8): 59.
[14] Ozawa D,Nakamura T,Koike M,etal.Shuttling protein nucleolin is a microglia receptor for amyloid beta peptide 1-42[J].Biol Pharm Bull,2013,36(10): 1587-1593.
[15] Bates KA,Verdile G,Li QX,etal.Clearance mechanisms of Alzheimer’s amyloid-beta peptide: implications for therapeutic design and diagnostic tests[J].Mol Psychiatry,2009,14(5): 469-486.
[16] Crews L,Masliah E.Molecular mechanisms of neurodegeneration in Alzheimer’s disease[J].Hum Mol Genet,2010,19(R1): R12-R20.
[17] Espuny-Camacho I,Dominguez D,Merchiers P,etal.Peroxisome proliferator-activated receptor gamma enhances the activity of an insulin degrading enzyme-like metalloprotease for amyloid-β clearance[J.J Alzheimers Dis,2010,20(4): 1119-1132.
[18] Song F,Poljak A,Valenzuela M,etal.Meta-analysis of plasma amyloid-β levels in Alzheimer’s disease[J].J Alzheimers Dis,2011,26(2):365-375.
[19] Memczak S,Jens M,Elefsinioti A,etal.Circular RNAs are a large class of animal RNAs with regulatory potency[J].Nature,2013,495(7441): 333-338.
[20] Hansen TB,Jensen TI,Clausen BH,etal.Natural RNA circles function as efficient microRNA sponges[J].Nature,2013,495(7441):384-388.
[21] Bernardino L,Xapelli S,Silva AP,etal.Modulator effects of interleukin-1β and tumor necrosis factor-α on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice culture[J].J Neurosci,2005,25(29):6734-6744.
山西省科技厅青年科技研究基金资助项目(2014021038-7);山西省卫计委青年基金资助项目(201301056)
王丹琼,女,1981-06生,硕士,主治医师,E-mail:wdq0603@163.com
2017-04-21
R741.05
A
1007-6611(2017)06-0546-04
10.13753/j.issn.1007-6611.2017.06.008
猜你喜欢
中老年保健(2022年2期)2022-08-24
中老年保健(2022年1期)2022-08-17
世界最新医学信息文摘(2021年12期)2021-06-09
天津医科大学学报(2019年6期)2019-08-13
奥秘(2018年9期)2018-09-25
分析化学(2017年12期)2017-12-25
天津医科大学学报(2015年2期)2015-12-22
安徽医科大学学报(2015年9期)2015-12-16
医学研究杂志(2015年4期)2015-06-10
中国当代医药(2015年16期)2015-03-01
