
儿童Gitelman综合征1例报道并文献复习
2017-07-31 21:43王小军杜东海石凯丽
山西医科大学学报 2017年2期
王小军, 杜东海, 石凯丽
(1山西省儿童医院内分泌科,太原 030013; 2山西医科大学儿科医学系; 3山西省儿童医院神经内科; *通讯作者,E-mail:kailihappy@126.com)
儿童Gitelman综合征1例报道并文献复习
王小军1, 杜东海2, 石凯丽3*
(1山西省儿童医院内分泌科,太原 030013;2山西医科大学儿科医学系;3山西省儿童医院神经内科;*通讯作者,E-mail:kailihappy@126.com)
Gitelman综合征; 低血钾; 低血镁; 低尿钙
低钾血症是内分泌科和泌尿科医生常见的临床问题,病因有遗传性和获得性两种,常见原因有腹泻、呕吐或利尿剂滥用等,而部分可引起低钾血症的少见病的诊断较为困难,其中一种易被忽视的疾病就是Gitelman综合征(Gitelman syndrome,GS)。GS又被称为家族性低钾低镁血症,是一种常染色体隐性遗传性肾小管疾病,其特点为低钾血症、低镁血症、代谢性碱中毒、低尿钙,血压正常或偏低[1]。该病自Gitelman等[2]在1966年首次发现以来,逐渐引起了临床医生的关注,但由于GS的发病率较低,国内相关报道较少。本文就我院收治的1例诊断为Gitelman综合征患者的临床资料进行回顾性分析,为该病的临床诊治提供参考,现报告如下。
1 临床资料
1.1 一般资料
患儿,女性,6岁,以“发热伴咳嗽、呕吐4 d”为主诉于2016年3月11日入院。入院4 d前患儿出现发热,最高体温39 ℃,热峰3次/d,伴持续性咳痰,痰量少,伴呕吐,2-3次/d,呈非喷射性,呕吐物为胃内容物,当地医院住院期间化验血钾波动在1.63-2.51 mmol/L之间,予输注头孢孟多酯、静脉补钾等治疗2 d无好转,为求进一步诊治入住我院。患儿自发病以来,精神可、食欲差,大便质稀,小便外观正常,无肢体抽搐,无多饮多食多尿史,否认服用利尿剂及其他药物史。智力运动发育正常,家族中无类似疾病史,父母亲均健康,无其他兄弟姐妹。入院查体:体温37 ℃,血压117/78 mmHg,甲状腺不大,双肺呼吸音粗,未闻及干湿性啰音,心、腹查体无异常,无手足麻木感,Trousseau征及Chvostek征均阴性。
1.2 诊疗经过
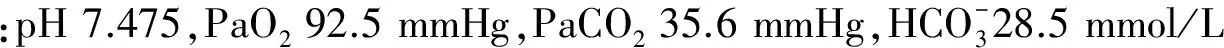
2 讨论
由于GS与Bartter综合征(Bartter syndrome,BS)二者的临床表现及实验室检查均相似,故以往将GS归为Bartter综合征的一个特殊类型,随着研究的深入,发现GS与BS的基因突变位点不同,是一种新的疾病。GS的病因主要是编码噻嗪类利尿剂敏感的Na+/Cl-共同转运体(NCCT)的SLC12A3基因发生突变。导致BS的突变基因包括SLC12A1、KCNJl、CLCNKB、BSND、CASR等5种[2]。BS在任意年龄段均可发病,但以5岁前儿童多见;GS则多在成年或青春期发病,儿童患者少见[3]。白种人中GS的发病率为1/40 000[4],且杂合子携带率约为1%[5]。日本人群中的发病率高达10.3/10 000[6]。
2.1 病理生理
2.1.1 电解质和代谢紊乱 NCCT的主要功能是介导肾脏远曲小管Na+和Cl-的重吸收,该转运体共由12个跨膜结构域组成,且氨基端和羧基端均位于细胞内。位于16号染色体长臂上的SLC12A3基因突变导致其编码的NCCT结构和/或功能异常,造成远曲小管中Na+和Cl-的重吸收障碍,引起Na+和Cl-丢失、血容量减少,导致血压下降,从而激活肾素-血管紧张素-醛固酮(RASS)系统,血浆肾素活性和醛固酮水平升高,作用于Na+/K+、Na+/H+通道促进Na+的重吸收、K+和H+的分泌,引起低血钾和代谢性碱中毒[7]。通过肠道吸收的Mg2+经肾小球滤过,在近端小管可重吸收10%,在髓袢升支经细胞旁途径通过被动扩散可重吸收50%-70%。血Mg2+下降可能与醛固酮作用有关,因管腔侧Na+重吸收增加形成了管腔侧负电位,并通过Mg2+/Na+交换的增多而增加尿Mg2+的量,造成血Mg2+降低[7]。虽然Ca2+也经肾小球滤过,但其转运途径与Mg2+不同;Ca2+在近端小管和髓袢升支处的被动重吸收主要由Na+/Cl-转运介导的跨膜梯度和细胞旁路途径介导,而Ca2+只在甲状旁腺激素和维生素D的作用下通过远曲小管进行主动转运。因Na+/Cl-联合转运功能的异常会使细胞内Cl-的超极化作用减弱,Ca2+回吸收增加,引起尿Ca2+减少[8]。本研究中患儿进食正常后仍有低血钾、低血镁,肾素活性增高,低尿钙等表现,考虑与上述作用机制有关。
2.1.2 肾活检 GS在肾脏最主要的病理改变是肾小球旁器增生,其原因为肾小球旁器在肾素的作用下,入球小动脉的平滑肌细胞转化为肾素分泌细胞而引起。但Shaer等[9]发现活检结果并不一致,且许多病理改变非GS所特有,故认为肾活检对GS诊断的参考价值有限。肾小球旁器的增生也可出现在慢性低血容量伴低钾血症的患者中,如长期应用催吐药和缓泻剂、家族性氯泻、利-范二氏综合征、BS等情况。而肾脏病理活检未见到肾小球旁器增生也不能排除GS[10]。而BS的肾活检也存在肾小球旁器肥大,故肾活检不能作为鉴别GS与BS的手段。
2.1.3 基因筛查 对GS的明确诊断有赖于基因检测。GS是一种常染色体隐性遗传性疾病,目前发现的SLC12A3基因突变超过250种。突变类型包括无义突变、错义突变、移码突变、缺失、插入和剪切位点突变等,中国人SLC12A3基因突变类型以T60M为主[3]。Lee等[11]报道部分GS患者同时存在SLC12A3基因和CLCNKB基因的突变,Enríquez等[12]报道了1例由CLCNKB基因p.A204T突变引起的GS,故当GS患者没有检测出SLC12A3基因突变时,应该进一步对其进行CLCNKB基因的检测。基因型或SLC12A3基因的突变数目不能预测疾病的严重程度或对治疗的反应[11]。
2.2 临床特点
GS与BS均存在低血钾、代谢性碱中毒、高肾素、高醛固酮血症和血压正常等特点。GS最突出的实验室检查为低镁血症和低尿钙[7];而BS则无低镁血症,且尿钙可正常或升高,故该两项指标可作为二者临床鉴别的主要依据。GS也常伴有轻至中度的低磷血症[13]。也有GS出现严重低磷血症和严重低钠血症的报道[14,15]。低血钾会引起乏力、心律失常、便秘、多尿和夜尿增多等。低钾引起的细胞内复极化改变是导致乏力的主要原因。长期低钾可引起肾小管上皮细胞的破坏及间质纤维化,从而出现多尿及夜尿增多等。但多数患者6岁前临床症状不典型,一般症状有肌无力、肢体麻木、手足搐搦等,还可伴有腹痛、呕吐、发热等。部分患者也可出现软骨钙质沉积,致使受累关节肿胀疼痛,多认为与低血镁有关。本患儿以发热、咳嗽、呕吐起病,考虑感染使电解质紊乱及酸碱水平失衡,进而使呕吐加剧,且患儿无肌肉症状,提示儿童患者症状可不典型,需及时监测相关检验指标。患儿病情好转后,虽然其家属拒绝进一步行肾活检及基因检测,但根据其低血钾、低血镁、肾素活性增高、低尿钙,且血钙及血压正常等特点,仍临床诊断GS。
2.3 治疗及预后
因GS病情较BS轻,故预后相对较好。目前GS尚无法完全治愈,只能通过补钾、补镁对症治疗以缓解临床症状,联合使用肾素抑制剂可维持电解质平衡,且无不良反应[16]。而联用保钾利尿剂雷米普利或安体舒通也可稳定维持血钾、血镁的水平[17]。目前关于GS患者的随访报道较少。Tseng等[18]对117例存在SLC12A3基因突变的GS患者给予补钾、补镁治疗,并进行了长期随访,在不服用中药或肾毒性药物的情况下,7例男性在平均随访12年时出现了慢性肾衰;4例男性和1例女性在平均随访11个月时发现了糖尿病;以上并发症与普通人群相比,发病时间提前且发病率较高。本患儿随访7个月,血钾上升至正常水平,且未再出现类似症状,其长期预后仍有待于跟踪随访。
综上,对于临床上遇见的低钾血症患者,尤其是症状不典型的儿童患者,要详细询问病史,同时也需要详询家族史,完善相关检查,降低漏诊及误诊率,早期诊断,合理治疗,以期改善患者预后。
[1] Graziani G,Fedeli C,Moroni L,etal.Gitelman syndrome:pathophysiological and clinical aspects[J].QJM,2010,103(10):741-748.
[2] Gitelman HJ,Graham JB,Welt LG.A new familial disorder charaeterized by hypokalemia and hypomagnesemia[J].Trans Assoc Am Physicians,1966,79:221-235.
[3] 陈占玲,何灵杰,张秀薇,等.一例Gitelman综合征患者临床资料与SLC12A3基因突变分析研究[J].中华内分泌代谢杂志,2015,31(10):910-912.
[4] Knoers NV,Levtchenko EN.Gitelman syndrome[J].Orphanet J Rare Dis,2008,3(22):1-6.
[5] Melander O,Orho-Melander M,Bengtsson K,etal.Genetic variants of thiazide-sensitive NaCl-cotransporter in Gitelman’s syndrome and primary hypertension[J].Hypertension,2000,36(3):389-394.
[6] Tago N,Kokubo Y,Inamoto N,etal.A high prevalence of Gitelman’s syndrome mutations in Japanese[J].Hypertens Res,2004,27(5):327-331.
[7] Pantanetti P,Arnaldi G,Balercia G,etal.Severe hypomagnesaemia-induced hypocalcaemia in a patient with Gitelman’s syndrome[J].Clin Endocrinol(Oxf),2002,56(3):413-418.
[8] Kurtz I.Molecular pathogenesis of Bartter’s and Gitelman’s syndromes[J].Kidney Int,1998,54(4):1396-1410.
[9] Shaer AJ.Inherited primary renal tubular hypokalemic alkalosis:a review of Gitelman and Bartter syndromes[J].Am J Med Sci,2001,322(6):316-332.
[10] 秦岭,邵乐平,任红,等.Gitelman综合征的表型特点及性别因素对表型的影响[J].中华肾脏病杂志,2009,25(7):532-537.
[11] Lee JW,Lee J,Heo NJ,etal.Mutations in SLC12A3 and CLCNKB and their correlation with clinical phenotype in patients with Gitelman and Gitelman-like Syndrome[J].J Korean Med Sci,2016,31(1):47-54.
[12] Enríquez R,Adam V,Sirvent AE,etal.Gitelman syndrome due to p.A204T mutation in CLCNKB gene[J].Int Urol Nephrol,2010,42(4):1099-1102.
[13] Viganò C,Amoruso C,Barretta F,etal.Renal phosphate handling in Gitelman syndrome—the results of a case-control study[J].Pediatr Nephrol,2013,28(1):65-70.
[14] Ali A,Masood Q,Yaqub S,etal.A case of Gitelman syndrome with severe hyponatraemia and hypophosphataemia[J].Singapore Med J,2013,54(1):e18-20.
[15] Guedes-Marques M,Silva C,Ferreira E,etal.Gitelman syndrome with hyponatremia,a rare presentation[J].Nefrologia,2014,34(2):266-268.
[16] 卢婉,郭颖,倪昶,等.青少年Gitelman综合征一例的临床表现及基因诊断分析[J].中华内分泌代谢杂志,2015,31(5):438-442.
[17] António JC,Alexandra C.Gitelman or Bartter type 3 syndrome A case of distal convoluted tubulopathy caused by CLCNKB gene mutation[J].BMJ Case Rep,2013,2013(1):1-4.
[18] Tseng MH,Yang SS,Hsu YJ,etal.Genotype,phenotype,and follow-up in Taiwanese patients with salt-losing tubulopathy associated with SLC12A3 mutation[J].J Clin Endocrinol Metab,2012,97(8):E1478-E1482.
王小军,男,1972-06生,主治医师,E-mail:wxj53488@163.com
2016-11-14
R591.1
B
1007-6611(2017)02-0196-03
10.13753/j.issn.1007-6611.2017.02.024
猜你喜欢
新疆大学学报(自然科学版)(中英文)(2022年4期)2022-08-02
中国生殖健康(2020年2期)2021-01-18
青岛大学学报(医学版)(2021年6期)2021-01-13
中华养生保健(2020年2期)2020-11-16
心肺血管病杂志(2019年12期)2019-05-20
小学生导刊(2018年13期)2018-06-29
中外医学研究(2018年5期)2018-04-10
中国生殖健康(2018年2期)2018-01-12
中学生物学(2016年12期)2017-04-06
现代检验医学杂志(2015年5期)2015-02-06
