
温度及压强对CO2-盐水系统界面张力的影响
2017-07-18 11:43:32季佳圆赵伶玲李偲宇
化工学报 2017年7期
季佳圆,赵伶玲,李偲宇
(东南大学能源热转换及其过程测控教育部重点实验室,能源与环境学院,江苏 南京 210096)
温度及压强对CO2-盐水系统界面张力的影响
季佳圆,赵伶玲,李偲宇
(东南大学能源热转换及其过程测控教育部重点实验室,能源与环境学院,江苏 南京 210096)
在超临界态CO2封存于深部盐水层过程中,温度、压强等控制条件是影响封存效率和封存量的重要因素。应用分子动力学模拟的方法对343~373 K和6~35 MPa范围内的CO2-NaCl盐水系统进行了界面张力(IFT)及界面特性的研究,分析了IFT随温度及压强的变化关系,并观测到了压力平衡点pplateau;从分子尺度(物质密度、界面过余量、界面水合物密度)分析了IFT随压强、温度的变化,以及pplateau产生的原因。结果表明,pplateau前压强升高或温度降低将导致CO2密度升高,IFT下降,而pplateau后IFT趋于稳定且受温度影响较小;CO2的界面过余量及界面处水合物数量随压强及温度变化,与IFT的变化相反;高压下界面水合物密度的饱和现象可能是pplateau产生的重要原因。
CO2-盐水系统;界面张力;分子模拟;温度;压强;水合物
引 言
深部盐水层CO2地质封存所需的注射能耗及最大地质埋存深度与 CO2-盐水之间的界面张力(interfacial tension,IFT)直接相关[1-4],并受温度和压强的制约。开展温度、压强对CO2-盐水间界面张力的影响研究,不仅可以分析IFT随温度、压强等控制参数的变化规律,还能阐述温度、压强对IFT产生影响的内在机理进而对指导不同环境条件(温度、压强)下的CO2地质封存设计,提高注射安全性及存储容量具有重大意义。
目前实验[1,5-8]已测定相关储层条件下 CO2-水和CO2-盐水系统的IFT值,并观测到IFT在定温条件下会随着压强升高而降低,并在压力平衡点pplateau之后趋于稳定值。实验还发现pplateau的大小与盐的种类及盐度无明显联系,仅随温度升高而上升。Chalbaud等[6]将pplateau的存在归因于CO2溶解度的影响,但尚未展开深入分析。
分子动力学模拟(molecular dynamics simulation,MD模拟)可以研究多相界面系统的微观特性,目前该方法已成功模拟了 CO2-水及 CO2-盐水系统,可获得与实验一致的IFT值[9-13],并能观测界面的微观现象,是一种有效的研究手段。
本文应用 MD模拟方法,对 343~373 K和6~35 MPa范围内的CO2-NaCl系统进行计算,分析了体相及界面各物质性质随环境条件的变化规律,包括CO2的密度、CO2的界面过余量、界面处CO2水合物数量等,探讨了IFT对温度及压强依赖关系的物理机理,尤其对pplateau现象的产生原因进行了分析,可为IFT的控制和预测提供理论依据。
1 研究对象及方法
1.1 对象及模型
本文根据Chalbaud等[6]对CO2-NaCl系统大范围温度及压强下的IFT实验研究结果,选择了温度及压强范围为 343~373 K和 6~35 MPa的CO2-NaCl系统为研究对象。具体工况参数列于表1。

表1 CO2-NaCl系统的温度和压强条件Table 1 Temperature and pressure conditions of CO2-NaCl system
在计算过程中,综合考虑了系统内分子间非键结作用力(范德华力、库伦静电力)及分子内键结作用力(键拉伸和键弯曲)。分别采用Lennard-Jones势能函数[14]模拟范德华力,库仑定律模拟库仑静电力,具体分子间势能函数如式(1)所示

其中,rij为原子i与j之间的距离;εij为势能阱的深度,εij(εiiεjj)1/2;σij为两体互相作用的势能为零时的距离,σij(σiiσjj)1/2;ε0为真空介电常数;qi及 qj为原子 i与j所带电荷量。其中采用PME技术[15]模拟分子间长程库仑作用力,范德华作用截距设定为0.9 nm[16-18]。此外,本文采用谐波势能函数[5]模拟键拉伸和键角弯曲等分子内键结作用力。
本文计算中,水分子选择柔性F3C模型[19],CO2选择柔性EPM2模型[20],盐离子采用Chandrasekhar等[21]开发的模型。应用MD软件Gromacs4.5[22]并采用周期性边界条件进行计算,所建立的横截面4 nm×4 nm的计算域示于图1。计算域中间区域为盐水,包括4323个水分子、147个Na+和147个Cl-,对应盐度为1.89 mol·L-1,两侧分别为 732个CO2分子。利用 Berendsen方法[23-24]来实现温度和压强的设定。由于系统在NPzT系综下20 ns达平衡态,故模拟时间运行30 ns,选取最后5 ns为有效数据进行分析。

图1 CO2-NaCl系统平衡状态Fig.1 CO2-NaCl solution system in equilibrium state
1.2 数据处理
计算系统中包括两个界面,根据界面张力γ定义[25],系统中γ可由式(2)计算

式中,pxx, pyy, pzz分别为沿x,y,z方向压强张量对z向的角分量。
吉布斯分界面(Gibbs dividing surface,GDS)为以体相为参照,垂直于界面方向上CO2相多余水分子与溶液相缺乏水分子相等的位置,其厚度采用0.1~0.9倍水密度之间的距离表示[26]。
界面过余量[27]表征物质在界面和体相中的量差异,本文中CO2-NaCl系统界面处i分量的界面总过余量可用式(3)表示

2 结果与讨论
2.1 IFT值的变化
本文分别模拟了343 K和373 K的各压强下盐浓度为1.89 mol·L-1的CO2-NaCl系统,IFT的计算结果示于图2。由图2可以看出,模拟所得的 IFT值与Chalbaud等[6]的实验数据吻合较好。在温度为343 K时[图2(a)],IFT值随着压强的不断升高,其减幅逐渐下降,最终在压力平衡点pplateau=15 MPa之后达到稳定值34 mN·m-1。373 K时[图2(b)]的IFT值变化与343 K时极为相似,IFT值同样随着压强的升高而下降,且下降速度不断趋于平缓,由于高压情况下实验设备等因素受限制,实验方法尚未得到25 MPa以上的数据点,故在实验压强范围内并未观测到压力平衡点。而本文应用分子模拟的方法[28],成功模拟了25~35 MPa下的CO2-NaCl系统,观测到压强在pplateau=25 MPa之后达到稳定值33 mN·m-1。

图2 343 K和373 K下CO2-NaCl系统的IFT值Fig.2 IFT values of CO2-NaCl system at 343 K and 373 K
对比图2(a)与图 2(b)还可以发现,当盐浓度一定(1.89 mol·L-1)时,pplateau之前的 CO2-NaCl系统任意压强点的IFT值在温度为343 K时均小于373 K;同时随着压强的不断升高,两者差值不断缩小;而至压力平衡点之后两温度下的 IFT稳定值相接近,故温度对高压系统IFT稳定值的影响不大。此外,由于343 K下压力平衡点为15 MPa,373 K压力平衡点为25 MPa,故可推测压力平衡点会随温度升高而增大。
2.2 密度分布图

图3 343 K和373 K下界面CO2、H2O、盐离子密度Fig.3 Density profiles of CO2, water and salts at 343 K and 373 K
为从分子角度更加详细地描述界面性质的变化,本文选取并计算了界面处 2 nm厚度区域的CO2、H2O、Na+及Cl-的密度,结果示于图3。由图3可以观测到:当从 CO2相向溶液相过渡时,CO2密度逐渐下降,H2O密度则逐渐上升;而盐离子密度则从界面中心向溶液相,由0逐渐达到某一稳定值。由图3可以看出,温度一定时,CO2在CO2相的密度随着压强的升高逐渐升高。由[图 3(a)]可以看出,温度为343 K时,当压强由6.5 MPa升高至22 MPa,CO2相的CO2密度从580 kg·m-3升高至800 kg·m-3左右;而温度为 373 K 时[图 3(b)],当压强由11 MPa升高至17 MPa,CO2相的CO2密度从约420 kg·m-3升高至 600 kg·m-3左右。此外,对比图3(a)和(b)还可知,当压强恒定在11 MPa时,温度由343 K升高到373 K,CO2相的CO2密度则由700 kg·m-3降至 420 kg·m-3左右。
综上所述,压强的升高和温度的降低均对界面处CO2相的CO2密度产生较大的影响,而CO2密度的变化与IFT值的变化联系密切。具体而言,压强升高或温度降低时,CO2分子排列紧凑,界面分子受力的不均匀性有所好转,增大了其与水分子之间的吸引力。高密度下CO2分子更易进入水相,从而破坏了部分水分子间的氢键作用,界面上水分子受到内部分子的引力有所降低。以上两方面因素导致了界面水分子所受内部吸引力的减小,进而使得IFT值下降。
2.3 CO2界面过余量
由上述有关界面处密度的讨论可知,温度及压强对CO2相的CO2密度有较大的影响,为了更加全面地分析界面处CO2分子对界面张力的影响,本文进一步分析了 343~373 K和 6~35 MPa的CO2-NaCl系统的 CO2界面过余量,计算结果示于图4。由图4可知,343 K和373 K时,CO2界面过余量均随着压强的升高而逐渐升高,直至到达稳定值。具体来说,在343 K时,CO2界面过余量随着压强的升高不断升高,从压强 6.5 MPa 的 1.24 μmol·m-2,直至压力平衡点 15 MPa之后达到并稳定在 1.50 μmol·m-2左右;373 K 时 CO2界面过余量变化规律与343 K时基本一致,界面过余量从压强8 MPa 时的 0.93 μmol·m-2开始逐渐升高,直至压力平衡点 25 MPa 后稳定在约 1.57 μmol·m-2。
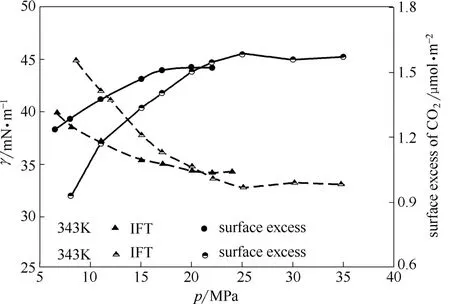
图4 343 K和373 K下的界面张力和CO2界面过余量Fig.4 IFT values and surface excess of CO2at 343 K and 373 K
由图4中CO2界面过余量及IFT和温度、压强的变化关系可以发现,在 pplateau之前,CO2界面过余量随着压强的升高或温度的降低而升高,而pplateau后,CO2界面过余量趋于稳定且受温度影响较小。CO2界面过余量受温度、压强的影响与IFT受温度、压强的影响结果恰好相反。这表明IFT受温度、压强变化的影响可理解为:压力平衡点之前的压强的升高或温度的降低均使得CO2分子在界面处的累积量增加,界面处CO2数量的增加可能是导致IFT降低的原因之一。
2.4 CO2水合物密度
由于CO2界面过余量随温度、压强变化的关系与IFT值变化规律相反,界面处CO2分子对IFT有着较大影响。考虑到界面附近CO2分子与水分子作用距离较近,极易形成CO2水合物[29-30],故本文进一步研究了343~373 K和6~35 MPa的CO2-NaCl系统界面处的水合物数量及密度。其中部分温度、压强条件下的界面处水合物分析结果示于图5。

图5 343 K和373 K下界面处水合物数量Fig.5 Hydrates quantities at 343 K and 373 K
由图5可知,当温度恒定时,界面处水合物数量随着压强的升高而增加,并逐渐趋于稳定值。当温度为343 K时,水合物数量由90不断升高并稳定至118(6.5~24 MPa);而373 K时,水合物数量则由74升高并稳定至130(11~35 MPa)。
为排除模型尺寸对水合物数量的影响,本文将各温度、压强条件下的水合物数量除以截面积,计算出水合物面密度,其值示于图6。
由图6可知,水合物密度随压强升高而逐渐降低,下降速率逐渐趋于平缓,最终在IFT的压力平衡点pplateau处达到饱和,密度值恒定。水合物密度与IFT值随温度、压强变化呈现相反趋势。因此可推测,温度及压强直接影响了界面处水合物密度,而界面处水合物密度的变化可能是导致界面张力变化的重要因素。
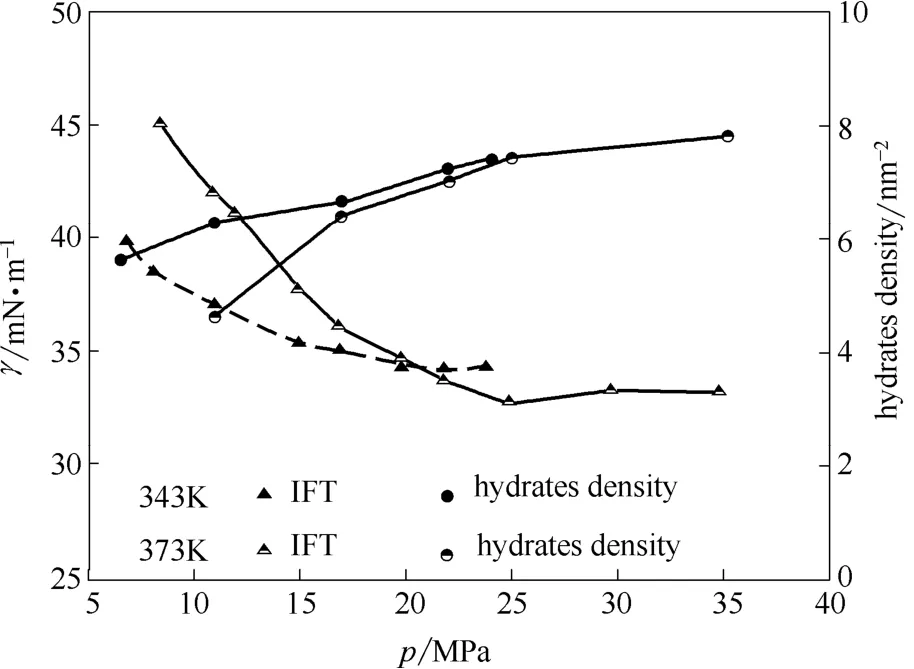
图6 343 K和373 K下的界面张力和界面处水合物密度Fig.6 IFI values and hydrates densities at 343 K and 373 K
3 结 论
本文应用MD模拟的方法进行了温度和压强在343~373 K 和 6~35 MPa范围内盐浓度为 1.89 mol·L-1的CO2-NaCl盐水系统的界面特性研究,探讨了IFT随温度、压强变化的微观机理及压力平衡点pplateau的存在原因,得出以下结论。
(1)CO2密度随着压强增加或温度降低而升高,其密度升高将增大CO2分子与界面水分子的引力,降低界面水分子所受到的水相内部的引力,最终导致压强增加或温度降低时IFT下降的趋势。
(2)恒温时 CO2界面过余量随压强升高而增加,并且在pplateau之后增加变缓。该值随温度、压强变化的趋势与IFT的变化趋势相反,CO2界面过余量对于IFT存在显著的负作用。
(3)界面处 CO2水合物的密度随温度、压强变化的趋势与IFT的变化趋势相反。温度恒定时,界面处水合物数量随着压强的升高而升高,升高速率逐渐降低,随后趋于定值。界面处水合物数量在高压下的饱和现象可能是导致压力平衡点pplateau产生的主要因素。
符 号 说 明
A ——界面截面积,m2
p ——压强,MPa
pplateau——压力平衡点,MPa
pxx——x方向压强张量对z向的角分量,Pa
pyy——y方向压强张量对z向的角分量,Pa
pzz——z方向压强张量对z向的角分量,Pa
qi——i离子的电荷数,e
qj——j离子的电荷数,e
rij——原子i和j之间的距离,nm
Si——i物质的界面过余量,μmol·m-2
T——温度,K
γ——界面张力,mN·m-1
εij——兰纳-琼斯势能阱深度,kJ·mol-1
ε0——真空介电常数
σij——兰纳-琼斯势能为零时距离,nm
[1] CHIQUET P, DARIDON J L, BROSETA D, et al. CO2/Water interfacial tensions under pressure and temperature conditions of CO2geological storage [J]. Energy Convers. Manage., 2007, 48 (3):736-744.
[2] JR F M O. Onshore geologic storage of CO2[J]. Science, 2009, 325(5948): 1656-1658.
[3] LI Z, DONG M, LI S, et al. CO2sequestration in depleted oil and gas reservoirs—caprock characterization and storage capacity [J]. Energy Convers. Manage., 2006, 47 (11/12): 1372-1382.
[4] JUNG J W, WAN J. Supercritical CO2and ionic strength effects on wettability of silica surfaces: equilibrium contact angle measurements[J]. Energy Fuels, 2012, 26 (9): 6053-6059.
[5] AGGELOPOULOS C A, ROBIN M, VIZIKA O. Interfacial tension between CO2and brine (NaCl+CaCl2) at elevated pressures and temperatures: the additive effect of different salts [J]. Adv. Water Resour., 2011, 34 (4): 505-511.
[6] CHALBAUD C, ROBIN M, LOMBARD J M, et al. Interfacial tension measurements and wettability evaluation for geological CO2storage [J]. Adv. Water Resour., 2009, 32 (1): 98-109.
[7] GEORGIADIS A, MAITLAND G, TRUSLER J P M, et al. Interfacial tension measurements of the (H2O+CO2) system at elevated pressures and temperatures [J]. J. Chem. Eng. Data, 2010, 55 (10): 4168-4175.
[8] HEBACH A, OBERHOF A, DAHMEN N, et al. Interfacial tension at elevated pressures-measurements and correlations in the water+carbon dioxide system [J]. J. Chem. Eng. Data, 2002, 47 (6): 1540-1546.
[9] ZHAO L L, LIN S C, MENDENHALL J D, et al. Molecular dynamics investigation of the various atomic force contributions to the interfacial tension at the supercritical CO2-water interface [J]. J.Phys. Chem. B, 2011, 115 (19):6076-6087.
[10] ZHAO L L, JI J Y, TAO L, et al. Ionic effects on supercritical CO2-brine interfacial tensions: molecular dynamics simulations and a universal correlation with ionic strength, temperature, and pressure [J].Langmuir, 2016, 32 (36): 9188-9196.
[11] DA-ROCHA S R P, JOHNSTON K P, WESTACOTT R E, et al.Molecular structure of the water-supercritical CO2interface [J]. J.Phys. Chem. B, 2001, 105 (48): 12092-12104.
[12] MÜLLER E A, MEJÍA A. Resolving discrepancies in the measurements of the interfacial tension for the CO2+H2O mixture by computer simulation [J]. J. Phys. Chem. Lett., 2014, 5 (7): 1267-1271.
[13] NIELSEN L C, BOURG I C, SPOSITO G. Predicting CO2-waterinterfacial tension under pressure and temperature conditions of geologic CO2storage [J]. Geochim. Cosmic. Acta, 2012, 81: 28-38.
[14] 范康年. 物理化学 [M]. 2版. 北京:高等教育社, 2005: 222.FAN K N. Physical Chemistry [M]. 2nd ed. Beijing: Higher Education Press, 2005: 222.
[15] AGGELOPOULOS C A, ROBIN M, PERFETTI E, et al. CO2/CaCl2solution interfacial tensions under CO2geological storage conditions:influence of cation valence on interfacial tension [J]. Adv. Water Resour., 2010, 33 (6): 691-697.
[16] KLAUDA J B, WU X, PASTOR R W, et al. Long-range Lennard-Jones and electrostatic interactions in interfaces: application of the isotropic periodic sum method [J]. J. Phys. Chem. B, 2007, 111(17): 4393-4400.
[17] DE LARA L S, MICHELON M F, MIRANDA C R. Molecular dynamics studies of fluid/oil interfaces for improved oil recovery processes [J]. J. Phys. Chem. B, 2012, 116 (50): 14667-14676.
[18] PATEL S A, BROOKS C L. Revisiting the hexane-water interface via molecular dynamics simulations using nonadditive alkane-water potentials [J]. J. Chem. Phys., 2006, 124 (20): 137-159.
[19] LEVITT M, HIRSHBERG M, SHARON R, et al. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution [J]. J. Phys. Chem. B, 1997, 101(25): 5051-5061.
[20] NIETO-DRAGHI C, DE BRUIN T, P REZ-PELLITERO J, et al.Thermodynamic and transport properties of carbon dioxide from molecular simulation [J]. J. Chem. Phys., 2007, 126 (6): 064509.
[21] CHANDRASEKHAR J, SPELLMEYER D C, JORGENSEN W L.Energy component analysis for dilute aqueous solutions of lithium(1+), sodium (1+), fluoride (1-), and chloride (1-) ions [J]. J. Am. Chem.Soc., 1984, 106 (4): 903-910.
[22] PRONK S, PALL S, SCHULZ R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit [J]. Bioinformatics, 2013, 29 (7): 845-854.
[23] BERENDSEN H J, POSTMA J P M, VAN GUNSTEREN W F, et al.Molecular dynamics with coupling to an external bath [J]. J. Chem.Phys., 1984, 81 (8): 3684-3690.
[24] BUSSI G, DONADIO D, PARRINELLO M. Canonical sampling through velocity rescaling [J]. J. Chem. Phys., 2007, 126 (1): 014101.
[25] ALEJANDRE J, TILDESLEY D J, CHAPELA G A. Molecular dynamics simulation of the orthobaric densities and surface tension of water [J]. J. Chem. Phys., 1995, 102 (11): 4574-4583.
[26] YUET P K, BLANKSCHTEIN D. Molecular dynamics simulation study of water surfaces: comparison of flexible water models [J]. J.Phys. Chem. B, 2010, 114 (43): 13786-13795.
[27] LI X, ROSS D A, TRUSLER J P, et al. Molecular dynamics simulations of CO2and brine interfacial tension at high temperatures and pressures [J]. J. Chem. Eng. Data, 2013, 117 (18): 5647-5652.
[28] LEACH A R. Molecular Modelling: Principles and Applications [M].2nd ed. England, Harlow: Prentice Hall, 2001.
[29] SADLEJ J, MAKAREWICZ J, CHAŁASIŃSKI G. Ab initio study of energy, structure and dynamics of the water-carbon dioxide complex[J]. J. Chem. Phys., 1998, 109 (10): 3919.
[30] TEWES F, BOURY F. Thermodynamic and dynamic interfacial properties of binary carbon dioxide-water systems [J]. J. Phys. Chem.B, 2004, 108 (7): 2405-2412.
Temperature and pressure effect on interfacial tensions of CO2-brine system
JI Jiayuan, ZHAO Lingling, LI Siyu
(Key Laboratory of Energy Thermal Conversion and Control of Ministry of Education, School of Energy & Environment,Southeast University, Nanjing 210096, Jiangsu, China)
Temperature and pressure are important effect factors of the efficiency and quantity of CO2storage in the deep saline aquifers. Molecular dynamics (MD) simulation is applied to investigate the CO2-NaCl systems in 343—373 K and 6—35 MPa, the interfacial tensions (IFT) which are obtained from the simulations are consistent with experimental results and the pressure balance point pplateauare observed as well in this paper. Meanwhile, the interfacial tension variations with the temperature and pressure are analyzed and the reasons of pplateaufrom the molecular viewpoint are explained. The results show that the pressure rise and temperature decline will increase CO2density and decrease IFT before pplateau, but after pplateauthe IFT will be stable and less affected by temperature.In addition, the changes of CO2surface excess and the hydrate quantities with temperature and pressure showed the opposite trend compared with IFT variations, the saturation phenomena of hydrates at the interface under high pressure may be the fundamental reason of pplateau.
CO2-brine system; interfacial tension; molecular simulation; temperature; pressure; hydrates
date:2016-12-30.
Prof. ZHAO Lingling, zhao_lingling@seu.edu.cn
supported by the National Natural Science Foundation of China (51106027).
TQ 021.2
A
0438—1157(2017)07—2880—06
10.11949/j.issn.0438-1157.20161837
2016-12-30收到初稿,2017-03-14收到修改稿。
联系人:赵伶玲。
季佳圆(1993—),女,硕士研究生。
国家自然科学基金项目(51106027)。
猜你喜欢
西南石油大学学报(自然科学版)(2021年3期)2021-07-16 05:27:08
科教新报(2021年11期)2021-05-12 19:50:11
烟台果树(2019年1期)2019-01-28 09:34:58
西南石油大学学报(自然科学版)(2018年6期)2018-12-26 01:00:14
传媒评论(2018年7期)2018-09-18 03:45:52
中国资源综合利用(2017年4期)2018-01-22 02:46:57
河北地质(2017年2期)2017-08-16 03:17:10
科学之谜(2016年9期)2016-10-11 08:59:04
IT时代周刊(2015年7期)2015-11-11 05:49:56
散文百家(2014年11期)2014-08-21 07:16:58
