
高效液相色谱法测定牙膏中的氨甲环酸
2017-06-07 08:23谭建华李慧勇王继才夏泽敏熊小婷刘家溍李燕飞
分析测试学报 2017年5期
杨 培,谭建华,李慧勇,王继才,夏泽敏,熊小婷,刘家溍,李燕飞
(广州质量监督检测研究院,广东 广州 510000)
高效液相色谱法测定牙膏中的氨甲环酸
杨 培*,谭建华,李慧勇,王继才,夏泽敏,熊小婷,刘家溍,李燕飞
(广州质量监督检测研究院,广东 广州 510000)
建立了丹磺酰氯柱前衍生/高效液相色谱测定牙膏中氨甲环酸(TA)的方法。牙膏经甲醇提取、氮吹至干,再用丹磺酰氯进行衍生。衍生物通过X-Bridge C18色谱柱(250 mm×4.6 mm×5 μm)分离,以乙腈和0.1%甲酸水溶液为流动相梯度洗脱,在紫外波长250 nm条件下进行定量测定。系统考察了缓冲溶液pH值、衍生温度和衍生时间对氨甲环酸衍生效率的影响。结果表明,在优化实验条件下,氨甲环酸衍生物与基体杂质达到有效分离,在1~425 mg/L范围内线性关系良好,相关系数为0.999 5;在20,200,1 600 mg/kg 3个加标浓度下的回收率为98.7%~102%,相对标准偏差为0.85%~2.5%,方法检出限为2.0 mg/kg。该方法准确、可靠、灵敏度高,适用于各类牙膏中氨甲环酸的测定。
氨甲环酸(TA);丹磺酰氯;牙膏;高效液相色谱法
氨甲环酸,又名凝血酸、止血环酸,是一种合成的氨基酸类抗纤溶制剂[1],其作用机制为通过抑制纤溶酶与纤维蛋白上赖氨酸的结合,降低纤溶酶降解纤维蛋白[2-3],从而达到止血、抗感染、促进上皮愈合与引导血管再生的功效[4-5]。1991年,栾庆先等[6]对含氨甲环酸牙膏的临床效果做了简单的测试和评价,结果表明使用含氨甲环酸牙膏后,患者的牙龈出血状况得到缓解。近期,行业调研结果显示,氨甲环酸作为缓解牙龈出血等症状的功效成分越来越广泛地被应用于牙膏的配方中。然而,氨甲环酸作为一种药物,在牙膏中过量添加可能会造成潜在危害。因此,为了促进企业更好的把控产品质量,保障消费者的健康安全,亟需建立牙膏中氨甲环酸的检测方法。
目前,针对氨甲环酸的测定方法包括气相色谱法[7]、气相色谱-质谱联用法[8]、高效液相色谱法[9-12]、薄层扫描法[13]、分光光度法[14]、液相色谱-质谱联用法[15]、荧光分析法[16-17]等。GB/T 32121-2015采用高效液相色谱-串联质谱法测定牙膏中氨甲环酸的含量,该方法灵敏度高,选择性强。但由于液质联用仪价格相对昂贵,仪器的线性范围较窄,以及牙膏中高浓度表面活性剂的基质干扰等问题限制了其应用推广。本研究采用丹磺酰氯柱前衍生/高效液相色谱建立了牙膏中氨甲环酸的测定方法,所生成的衍生物具有较理想的反相保留,且改善了氨甲环酸的紫外吸收特性,在增强定性可靠性的同时大大提升了方法灵敏度。
1 实验部分
1.1 仪器与试剂
高效液相色谱仪配置PDA二极管阵列检测器(美国Waters公司2695-2998),N-EVAP系列氮吹仪(美国Organomation公司),Milli-Q 超纯水器(美国Millipore公司),Allegra 64R centrifuge台式高速冷冻离心机(美国Beckman Coulter公司),电子分析天平(精度0.01 mg和0.1 mg),pH计。氨甲环酸标准品(纯度98%,上海Anpel公司),丹磺酰氯(纯度98%,美国赛默飞世尔科技有限公司),甲酸、乙腈(色谱纯,美国Spectrum公司),无水碳酸氢钠、氢氧化钠(分析纯,广州试剂厂),其他试剂均为分析纯,实验用水为超纯水。
1.2 标准溶液的制备
准确称取适量的氨甲环酸标准品(精确至0.01 mg),用甲醇溶液配成约为500 mg/L 的标准储备溶液,逐级稀释成浓度分别为0,1,10,20,40,80 mg/L 的氨甲环酸系列标准工作溶液。
1.3 样品的提取
称取0.5 g 牙膏样品(精确至0.1 mg)于10 mL 比色管中,甲醇定容,于涡旋振荡器上混匀1 min,超声提取10 min,静置至室温,得样品溶液。
1.4 衍生化
标准溶液衍生化:取500 μL 氨甲环酸标准工作溶液至10 mL 比色管中,40 ℃ 下氮气吹扫至干,再依次加入500 μL 丹磺酰氯溶液(1 mg/mL,丙酮作为溶剂)和500 μL NaHCO3缓冲溶液(100 mmol/L,pH 9.5),于涡旋振荡器上混匀1 min,50 ℃水浴保温3 min 后,混匀过滤,待测。
样品溶液衍生化:取500 μL 的上述样品溶液至10 mL 比色管中,其他衍生步骤与标准溶液衍生化步骤相同。
1.5 色谱条件
色谱柱:X-Bridge C18(250 mm×4.6 mm×5 μm);流速:1.0 mL/min;检测波长:250 nm;进样量:10 μL;柱温:30 ℃;流动相:乙腈(A)和0.1%甲酸水溶液(B),梯度洗脱程序:0~4 min,80% B;4~6 min,80%~57% B;6~20 min,57% B;20~22 min,57%~0% B;22~26 min,0% B;26~28 min,0%~80% B。
2 结果与讨论
2.1 检测波长的选择
在210~400 nm波长范围内对氨甲环酸衍生物溶液进行全波长扫描,氨甲环酸衍生物在250,320 nm处均有较大的紫外吸收,其中250 nm为其最大紫外吸收波长,且溶剂、基质的干扰较小,故选择250 nm作为检测波长。
2.2 衍生试剂的选择及条件优化
氨甲环酸(TA)极性较强,且紫外吸收为末端吸收。在分析测试过程中,方法灵敏度较低且易被牙膏中其他成分干扰。因此,本研究尝试采用柱前衍生法进行分析。目前针对氨基基团常用的衍生试剂有邻苯二甲醛、苯甲酰氯、丹磺酰氯等,其中丹磺酰氯具有衍生操作简单、衍生物稳定性好、荧光和紫外吸收较强、灵敏度高、反应范围宽、基体干扰不明显等优点[18-19]。因此本研究采用丹磺酰氯作为衍生试剂。
丹磺酰氯可与伯胺或仲胺基上的活泼氢反应,脱掉1分子的HCl生成具有荧光和紫外吸收的衍生物,具体的磺酰化反应式如下:

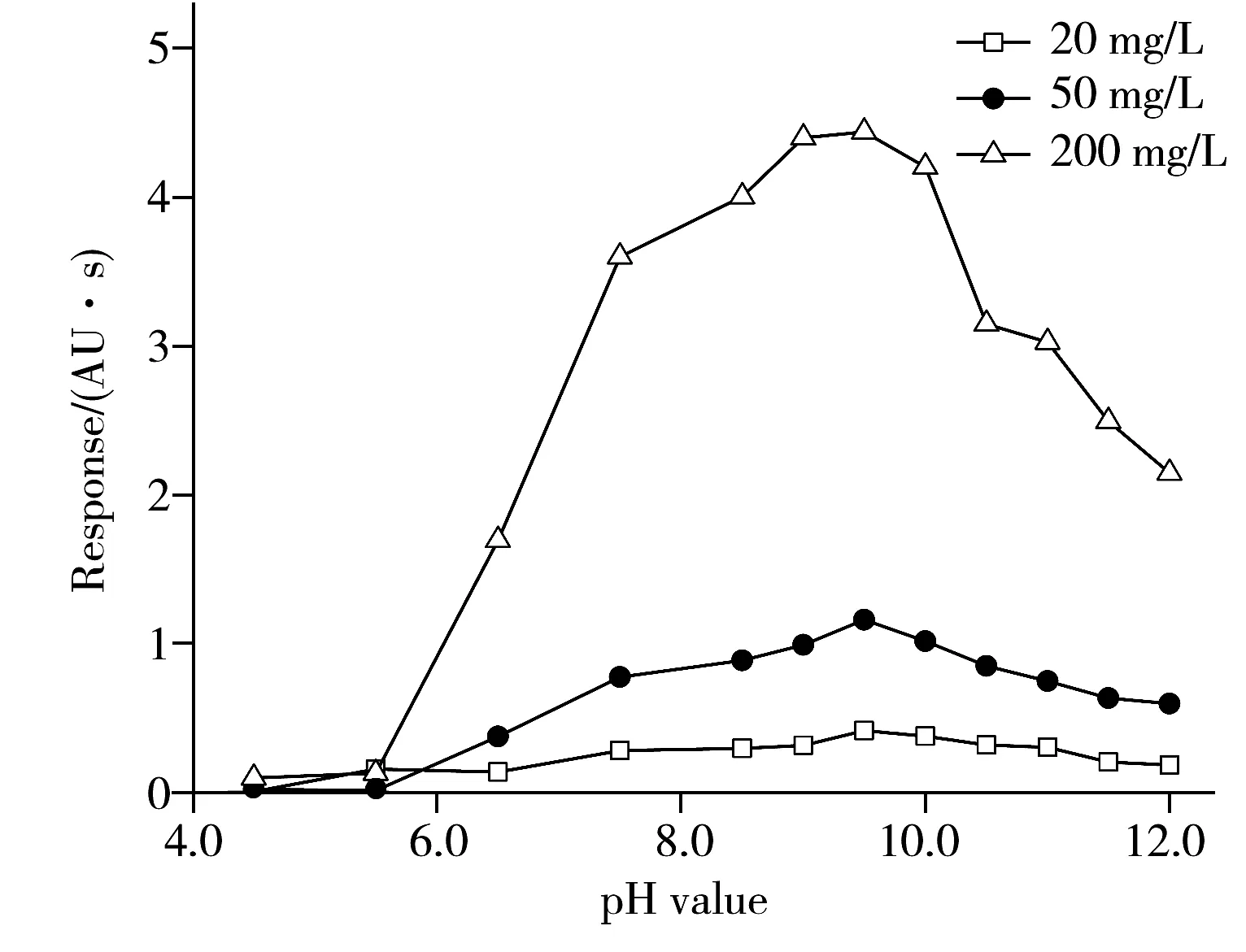
图1 缓冲溶液pH值对氨甲环酸衍生物响应面积的影响Fig.1 Effect of pH values of buffer solution on response area of TA derivative
在这一反应过程中起决定性作用的条件有缓冲溶液pH值、衍生温度和衍生时间[20]。因此本研究针对这3个影响因素对氨甲环酸的柱前衍生反应条件进行优化。2.2.1 缓冲溶液pH值的选择 选择NaHCO3缓冲溶液体系进行试验,比较了不同pH值(4.5~12.0)条件下氨甲环酸的衍生效率。分别采用10% H3PO4水溶液和1% 氢氧化钠水溶液调节NaHCO3溶液的pH值,其他反应条件与“1.4”一致。由图1可知,当缓冲溶液pH<9.5时,氨甲环酸衍生物的响应面积随着缓冲液pH值的增大而逐渐增大;当缓冲溶液pH>9.5时,氨甲环酸衍生物的响应面积随着缓冲液pH值的增大而逐渐减小。因此,本文选择缓冲溶液最佳pH值为9.5,此时氨甲环酸的衍生效率最佳。
2.2.2 衍生化温度的选择 比较了不同温度条件对氨甲环酸衍生效率的影响。结果显示,当衍生温度<50 ℃时,氨甲环酸衍生物的响应面积随着温度升高而逐渐增大;当衍生温度>50 ℃时,氨甲环酸衍生物的响应面积随着温度升高而逐渐减小。因此,本研究选用50 ℃作为氨甲环酸柱前衍生化的最佳温度。2.2.3 衍生化时间的选择 比较了不同衍生反应时间对氨甲环酸衍生效率的影响。结果显示,当柱前衍生化时间小于3 min时,随着反应时间的增加,氨甲环酸衍生物的响应面积略微增大,当柱前衍生化时间大于3 min时,继续延长反应时间,氨甲环酸衍生物的响应面积无明显变化。因此适宜的反应时间选择为3 min。
2.2.4 衍生物稳定性实验 将氨甲环酸衍生后的溶液分别放置不同时间(0 h,2 h,8 h,1 d,2 d,3 d),结果显示,3 d内氨甲环酸衍生物的响应面积基本不变,说明氨甲环酸衍生物在室温下3 d内稳定。
2.3 线性关系与检出限
配制浓度分别为1,10,20,40,80,200,425 mg/L的氨甲环酸系列标准溶液,在优化实验条件下对氨甲环酸衍生物进行测定,以质量浓度(ρ,mg/L)为横坐标,峰面积(A)为纵坐标绘制标准曲线。结果表明,在1~425 mg/L范围内,氨甲环酸衍生物的线性关系良好,线性方程为A=19 500ρ+78 200,相关系数(r2)为0.999 5。以信噪比S/N≈3的浓度作为检出限,S/N≈10 时的浓度作为定量下限,得到氨甲环酸的检出限和定量下限分别为2.0 mg/kg和7.0 mg/kg,方法的灵敏度可满足分析要求。
2.4 回收率与精密度
为了验证该方法对不同基质牙膏样品的适用性,选取3种不同配方体系的空白牙膏样品(水合硅石,磷酸氢钙和碳酸钙体系)作为研究对象,分别加入低、中、高3个浓度(20,200,1 600 mg/kg)的氨甲环酸标准溶液,进行加标回收率实验。结果显示,3个加标水平的回收率为98.7%~102%,相对标准偏差为0.85%~2.5%,表明方法具有较高的准确度,且精密度良好(见表1)。

表1 氨甲环酸标准溶液的回收率与相对标准偏差(n=5)Table 1 Recoveries and relative standard deviations(RSDs) of TA standard solution(n=5)
2.5 实际样品的分析
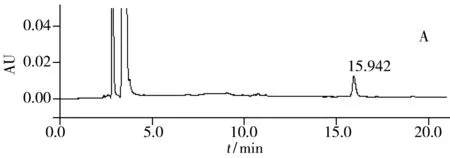
采用本方法对8支市售牙膏样品进行氨甲环酸含量的测定。结果显示,两支中草药牙膏检出氨甲环酸,其含量分别为0.073%和0.043%,标样谱图和实际样品谱图见图2。
3 结 论
本研究采用丹磺酰氯柱前衍生/高效液相色谱建立了牙膏中氨甲环酸的检测方法。对缓冲溶液的pH值、衍生温度和衍生时间等条件进行了优化,并将该方法应用于不同配方牙膏产品的检测。结果表明:氨甲环酸衍生物能够与杂质进行有效分离,不仅解决了目前检测方法干扰多、灵敏度低等问题,而且结果准确、灵敏、可靠。
[1] Zhang X L.ClinicalDrugReference.Chengdu:Sichuan Science and Technology Press(张象麟.药物临床信息参考.成都:四川科学技术出版社),2008:828-829.
[2] Tengborn L,Blombck M,Berntorp E.Thromb.Res.,2014,135(2):231-242.
[3] Simonazzi G,Saccone G,Berghella V.ActaObstet.Gyn.Scan.,2016,95(7):837.
[4] Rao F,Ding H,Wang Y,Chen F Y,Shi C H,Wang W S.ChongqingMed.(饶峰,丁浩,王炎,陈福宇,史晨辉,王维山.重庆医学),2016,45(9):1233-1235.
[5] Long G,Jiang J Q.Lab.Med.Clin.(龙刚,蒋俊强.检验医学与临床),2016,13(6):852-854.
[6] Luan Q X,Ouyang X Y,Zhang S W,Cao C F.Chin.J.Conservat.Dent.(栾庆先,欧阳翔英,张世卫,曹采方.牙体牙髓牙周病学杂志),1991,3(3):162-163.
[7] Almer S,Andersson T,Ström M.J.Clin.Pharmacol.,1992,32(1):49-54.
[8] Moore K A,Morin I,Marenco T,Lavigne J R,Morelli G.Am.J.Ther.,2012,19(3):190-198.
[9] Ying S,Wu K L,Sue J W,Kumar A S,Zen J M.J.Pharm.Biomed.Anal.,2008,48(5):1446-1450.
[10] Chen X Y,Qiu H,Wang M F,Zhang Q Q.Chin.J.Clin.Pharm.(陈娴瑛,邱涵,王美芳,张其清.中国临床药学杂志),2008,17(5):298-299.
[11] Liu C G.HeraldMed.(刘成广.医药导报),2007,26(10):1214-1215.
[12] Du N,Wang Y,Cai M M,Di B.Chin.Pharm.J.(杜宁,王玉,蔡美明,狄斌.中国药学杂志),2010,45(2):144-147.
[13] Hosiana B T,Endang S,Mochammad Y,Gunawan I.J.Liq.Chromatogr.Relat.Technol.,2005,28(20):3243-3254.
[14] Raza A.Anal.Lett.,2006,39(10):2217-2226.
[15] Bojko B,Vuckovic D,Cudjoe E,Hoque M E,Mirnaghi F,Wasowicz M,Jerath A,Pawliszyn J.J.Chromatogr.B,2011,879(32):3781-3787.
[16] Duangrat C,Wongsri K,Pongpaibul Y.Int.J.Cosmet.Sci.,2007,58(4):215-227.
[17] Wang X,Wang Q,Jiang A,Hu Y,Qiu X M,Li J H.Chin.J.Mod.Appl.Pharm.(王霞,王琼,江爱,胡焰,邱细敏,李健和.中国现代应用药业),2014,31(7):900-906.
[18] Molins-Legua C,Campíns-Falcó P,Sevillano-Cabeza A,Pedrón-Pons M.Analyst,1999,124(4):477-482.
[19] Dong W F,Li X Z,Lin W X.J.DalianInst.LightInd.(董伟峰,李宪臻,林维宣.大连轻工业学院学报),2005,24(2):115-118.
[20] Paleologos E K,Kontominas M G.Anal.Chem.,2004,76(5):1289-1294.
Determination of Tranexamic Acid in Toothpaste by High Performance Liquid Chromatography
YANG Pei*,TAN Jian-hua,LI Hui-yong,WANG Ji-cai,XIA Ze-min,XIONG Xiao-ting,LIU Jia-jin,LI Yan-fei
(Guangzhou Quality Supervision and Testing Institute,Guangzhou 510000,China)
A high performance liquid chromatographic(HPLC) method was developed for the determination of tranexamic acid(TA) in toothpaste.Sample was extracted with methanol, and the extract was evaporated to dryness under nitrogen before it was derived with dansyl chloride.Chromatographic separation was performed on an X-Bridge C18(250 mm×4.6 mm×5 μm) column using 0.1% formic acid-acetonitrile mixture aqueous solution as mobile phase.The ultraviolet detection wavelength was set at 250 nm.Effects of pH value of buffer solution,reaction temperature and time were investigated.Under the optimized experimental conditions,the TA derivative was successfully separated from the other impurities, and showed a good linearity in the range of 1-425 mg/L with a correlation coefficient(r) of 0.999 5.At three spiked concentrations of 20,200,1 600 mg/kg,the recoveries ranged from 98.7% to 102% with relative standard deviations(RSDs) of 0.85%-2.5%.The limit of detection for this method was 2.0 mg/kg.With the advantages of accuracy,reliability and high sensitivity,this method was suitable for the determination of tranexamic acid in different kinds of toothpaste.
tranexamic acid(TA);dansyl chloride;toothpaste;high performance liquid chromatography(HPLC)
2016-12-12;
2017-01-20
10.3969/j.issn.1004-4957.2017.05.015
O657.72;TQ657.3
A
1004-4957(2017)05-0665-04
*通讯作者:杨 培,硕士,工程师,研究方向:日化产品的分析测试,Tel:020-82022324,E-mail:yangpei0917@163.com
猜你喜欢
宁夏医学杂志(2020年3期)2021-01-21
分析化学(2019年3期)2019-03-30
当代医药论丛(2017年22期)2017-04-12
中华骨与关节外科杂志(2016年5期)2016-05-17
科技与创新(2016年5期)2016-03-17
化工生产与技术(2016年5期)2016-03-13
川北医学院学报(2015年5期)2015-12-07
山东工业技术(2015年6期)2015-07-27
