
1 例儿童静脉畸形骨肥大综合征的临床与基因突变分析
2017-06-05 15:11赵丽君田勇丽杨跃红寇敏
临床医药实践 2017年5期
赵丽君,田勇丽,杨跃红,寇敏
(山西省儿童医院,山西 太原 030013)
1 例儿童静脉畸形骨肥大综合征的临床与基因突变分析
赵丽君,田勇丽,杨跃红,寇敏
(山西省儿童医院,山西 太原 030013)
目的:探讨1 例静脉畸形骨肥大综合征(KTS)患儿的临床表现及其分子生物学基础。方法:收集患者的临床资料、实验室检查结果,抽取外周静脉血,提取基因组DNA,聚合酶链反应,测序确定突变情况。结果:患者临床表现、实验室检查符合KTS诊断。基因突变分析显示,患者AGGF1基因第6号外显子发生错义突变,为c.923A>G杂合突变,造成第923 位氨基酸由天冬酰胺改变为丝氨酸。蛋白序列的保守性分析及突变蛋白的功能分析,均支持该突变为致病突变。结论:通过临床及实验室检查,确诊1 例KTS患者,通过AGGF1基因突变分析,从分子遗传学方面证实患者KTS的诊断。
静脉畸形骨肥大综合征;基因突变;儿童
静脉畸形骨肥大综合征(KTS)是一种少见的先天性静脉畸形病变,1900年由法国医师Klippel和Trenaunay首次报道,其以血管畸形、骨肥大和软组织增生为主要临床表现[1]。本病好发于儿童及青少年,病程进展缓慢,病情复杂,并随年龄增长而加重,部分患者可伴有多趾、巨趾、并趾畸形及淋巴系统异常,下肢多见。目前本病的遗传方式不明确,多数认为AGGF1基因的变异与KTS相关[1]。本研究分析1 例KTS患者的临床、实验室检查和AGGF1基因突变情况,旨在探讨KTS患者的临床表现及分子生物学基础,以提高对本病的认识。
1 资料与方法
1.1 一般资料
患儿,女,7 岁,因血尿8 d于2016年7月就诊于山西省儿童医院。患儿8 d前无诱因出现肉眼血尿,尿液呈酱油色,无泡沫、少尿,无水肿,病初1 d伴低热,体温37.5 ℃左右,有轻咳,无痰,无皮疹、关节肿痛,无头痛、头晕,无恶心、呕吐、尿频、尿急、尿痛等症状,在当地医院诊断“急性肾小球肾炎”,输液治疗7 d(具体用药不详)病情无明显好转,为求进一步诊治入住我科。既往史、个人史、家族史无特殊。入院查体:体温37 ℃,脉搏92 次/min,呼吸20 次/min,血压121/54 mm Hg(1 mm Hg=0.133 kPa),神清,反应可,四肢、颈部、背部、臀部均可见大片状红色斑疹,浅表淋巴结未触及肿大,咽充血,双肺呼吸音粗,未闻及干湿啰音,心音有力,律齐,腹软,肝脾肋下未及,双肾区叩痛阴性,移动性浊音阴性,肠鸣音正常。右手拇指背面可见一花生大小突起皮面的赘生物,右手中指、食指粗大,中指外翻(见图1)。入院诊断:血尿原因待查,急性肾小球肾炎?上呼吸道感染。经皮肤科会诊,补充诊断:骨肥大静脉曲张痣综合征。
经患者知情同意,山西省儿童医院伦理委员会同意,抽取外周血,提取基因组DNA进行基因分析。
1.2 基因分析方法
1.2.1 基因组DNA提取

图11 例静脉畸形骨肥大综合征患儿的临床表现
采集患儿静脉血2 mL,置乙二胺四乙酸(EDTA)抗凝管中,用DNA提取试剂盒(康维世纪,北京)提取外周血白细胞基因组DNA。
1.2.2 聚合酶链反应(PCR)
1.2.2.1 引物设计
使用UCSC数据库查找AGGF1基因序列,试用Primer3.0进行引物设计,设计完成后进行序列合成,扩增AGGF1基因的14 个外显子及其与内含子的交界处,引物由北京迈基诺基因科技股份有限公司合成。AGGF1基因扩增引物(见表1)。
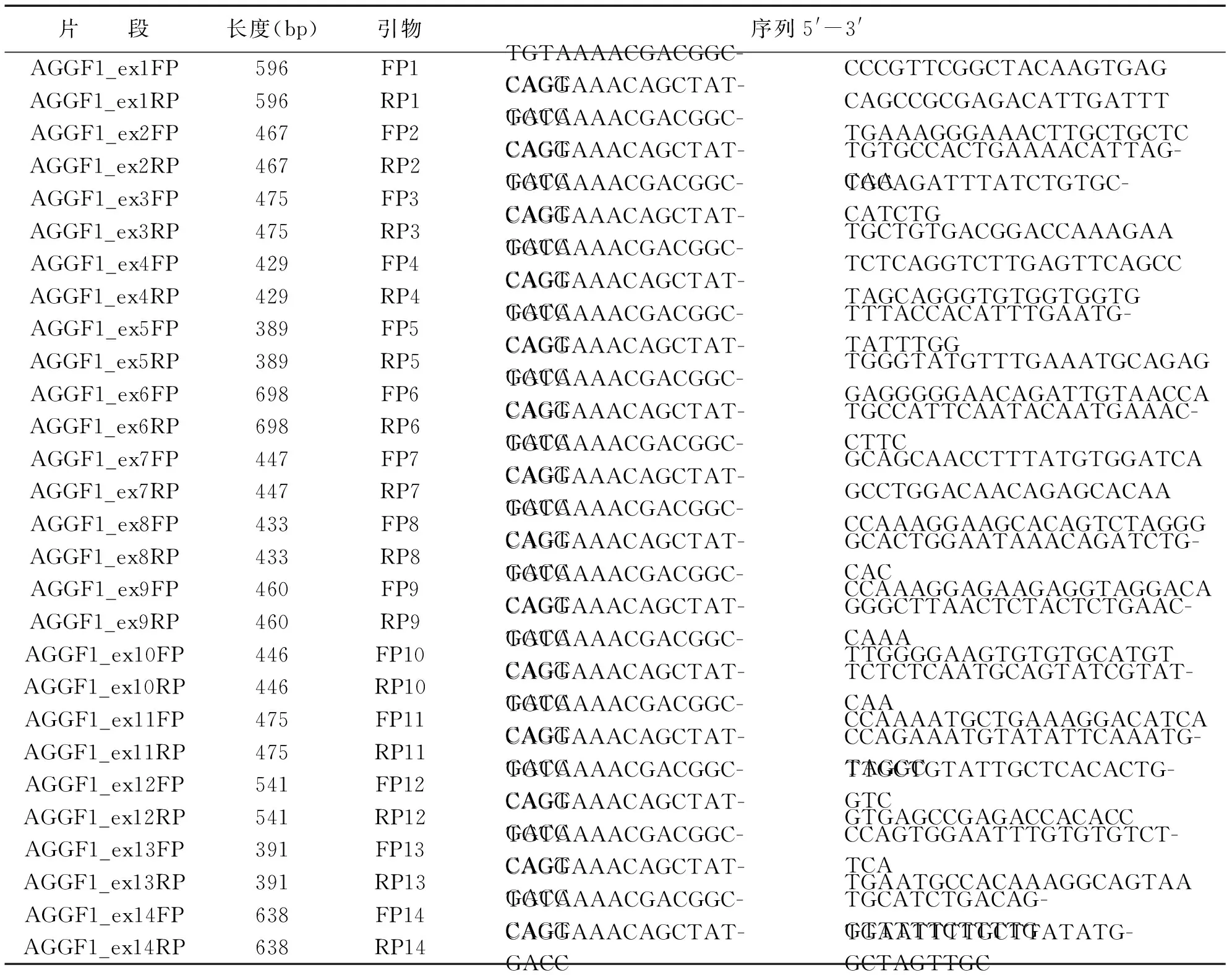
表1 AGGF1基因扩增引物
1.2.2.2 PCR扩增条件
95 ℃预变性60 s,94 ℃变性30 s,60 ℃退火30 s,72 ℃延伸45 s,循环35 次;后72℃延伸5 min,循环反应结束后取4 μL PCR产物经1.5%琼脂糖凝胶电泳鉴定PCR产物片段大小。
1.2.3 DNA测序
采用DNA纯化回收试剂盒(北京迈基诺基因科技股份有限公司)纯化回收PCR产物。再进行上机前PCR,采用之前设计好的引物,PCR反应条件为:96 ℃预变性1 min 30 s;96 ℃变性12 s,50 ℃退火6 s,60 ℃延伸3 min 20 s,循环25 次。反应结束后取PCR产物,送北京迈基诺基因科技股份有限公司用ABI3130荧光自动测序仪(ABI,美国)测序。
1.2.4 测序结果分析
测序结果与人类参考基因组(hg19)进行比对分析,确定突变位点及突变碱基。然后通过ANNOVER软件对突变位点进行注释,注释内容包括dbSNP数据库、HGMD数据库和正常人数据库1 000 Genomes、ESP6 500和ExAC等。根据注释信息排除多态性位点,确定其是否为已报道致病突变或新发突变位点。
1.2.5 对突变蛋白的功能预测
首先分析突变位点在不同物种中的保守性,其次使用非同义突变功能预测软件SIFT[2]、Polyphen-2[3]、Mutation Taster[4]对含错义突变的AGGF1蛋白进行致病性预测。
2 结 果
2.1 实验室和影像学检查结果
血系列:白细胞计数4.15×109/L,中性粒细胞0.436,淋巴细胞0.388,单核细胞0.145,嗜酸性粒细胞0.029,红细胞计数4.0×1012/L,血红蛋白浓度122 g/L,血小板计数222×109/L,C反应蛋白0.5 mg/L。生化:丙氨酸氨基转移酶12 U/L,天冬氨酸氨基转移酶31 U/L,γ-谷氨酰转肽酶10 U/L,白蛋白31.7 g/L,球蛋白25.8 g/L,尿素氮3.1 mmol/L,肌酐53 μmol/L,总胆固醇3.66 mmol/L,电解质正常。凝血功能:凝血酶原时间14.0s,纤维蛋白原3.94 g/L,活化部分凝血活酶时间31.7 s,凝血酶时间18.9 s。免疫球蛋白G(IgG)10.4 g/L,免疫球蛋白A(IgA)3.63 g/L,免疫球蛋白M(IgM)1.08 g/L,总免疫球蛋白E(IgE)26.9kIU/L,补体C31.06 g/L,补体C40.234 g/L。淋巴细胞亚群:T细胞68.9%,辅助性T细胞(Th)35.1%,抑制性T细胞(Ts)29.0%,Th/Ts 1.21,自然杀伤(NK)细胞4.8%,细胞因子诱导的杀伤(CIK)细胞2.4%,B细胞23.0%。血沉45 mm/h。肺炎支原体抗体阴性,院感九项正常。尿常规为:酸碱度6.56,蛋白质阴性,红细胞200.00个/μL,尿红细胞位相:蛋白(+-),红细胞数目>100个/HPF,变形60%,形状草莓、面包圈、穿孔,24 h尿蛋白定量0.34 g/24 h。肾早期损伤检测:尿IgG 14.5 mg/L,尿微量白蛋白87.4 mg/L,尿转铁蛋白4.58 mg/L,尿α1微球蛋白5.59 mg/L,便常规正常。胸片:双肺纹理粗多紊乱。腹部彩超:双肾大,与原发病有关。胃肠道彩超:腹腔内未见明显异常回声。
2.2 基因突变分析结果
2.2.1 PCR扩增产物检测
以患者DNA为模板,分别用引物进行PCR扩增,产物在1.5%琼脂糖凝胶电泳EB染色后,均见一条清晰条带,且与预期片段大小相符(见图2)。

图2 AGGF1扩增后1.5%琼脂糖凝胶电泳检测
2.2.2 基因突变分析
测序结果显示,患者的AGGF1基因第6号外显子发生错义突变,为c.923A>G杂合突变,造成第923位氨基酸由天冬酰胺突变为丝氨酸,即p.N308S(见图3),测序结果与人类参考基因组(hg19)比对,再通过ANNOVER软件对突变位点进行注释,表明c.923A>G不是多态性位点,确定其为未报道过的新突变。突变蛋白的功能预测:AGGF1蛋白923位的天冬酰胺在人,大鼠,小鼠,兔,牛,马,荷兰猪,恒河猴,狗中高度保守,同时SIFT、Polyphen-2和Mutation Taster对错义突变的功能影响预测结果:该突变可能影响AGGF1蛋白的功能,是致病性突变。SIFT、Polyphen-2和Mutation Taster评分分别为1、0.001和1。
3 讨 论
KTS是一种以静脉畸形合并其他多器官畸形的先天性疾病,在起病早期由于症状不典型,常被误诊为“胎记”“血管瘤”“静脉曲张”等疾病,延误了正常的诊治,导致患儿成长后残疾,因此目前已引起学者们的高度重视。在KTS三联征中,一般以血管瘤(痣)最先出现者为多,多在出生时即有或在出生后不久出现,以后随年龄增长,逐渐出现浅静脉曲张和骨、软组织增生。本病累及内脏及颅内血管时,还可出现膀胱、脾、结肠、肝脏等内脏血管畸形,可表现为消化道出血、泌尿道出血、脑出血。Husmann等[5]报道KTS患者中30%泌尿生殖系统受累,其中7%累及生殖器皮肤,7%累及泌尿生殖内脏器官,16%二者均累及。9%的KTS患者出现肉眼血尿,分别来自膀胱、尿道及肾脏。本例患儿急性起病,以肉眼血尿为主要表现,伴轻度蛋白尿,化验尿红细胞位相:红细胞>100个/HPF,变形60%,24 h尿蛋白定量0.34 g/24 h,诊断急性肾小球肾炎。本病易合并血管畸形,故未行肾活检。给予阿魏酸哌嗪、卡托普利、百令胶囊治疗后,肉眼血尿明显减轻,出院后2 个月复查尿常规、肾早期损伤检测等均正常。KTS的病因及发病机制目前尚不清楚[6],多数认为与先天性中胚层血管发育异常、遗传及基因突变有关。由于胎儿时期发育成血管和软组织的中胚层发育异常,导致相应部位的肢体浅静脉数量增多,管径扩大和血流增加;深静脉发育细小、闭塞或瓣膜缺如[7],进而肢体血容量持续增多造成骨骼和软组织过度生长[8],引起患肢一系列临床表现。研究表明AGGF1基因的变异与KTS相关,AGGFl基因全长34 kb,由14 个外显子组成,编码714 个氨基酸,所表达的蛋白相对分子量为81 kD,AGGF1基因编码一个强效血管生成因子,可以促进血管内皮细胞的增殖。KTS相关的基因突变导致血管生成因子活性增强,这是形成KTS病理改变的分子基础[7]。本例患者行基因检测明确为AGGF1基因第6号外显子发生错义突变,为c.923A>G杂合突变,造成第923 位氨基酸由天冬酰胺改变为丝氨酸。通过对不同种属该位点的蛋白序列的保守性分析及多种点突变功能预测软件分析,均支持该突变为致病突变。本例患者的突变属于新突变,其父母均检测到AGGF1基因的杂合突变,但未发病,结合类似研究报道,我们认为KTS是一类具有遗传基础的先天性疾病,AGGF1基因可能不是该例病患发病的唯一突变基因,KTS发病可能有其他基因作为致病基因,也可能在整个血管母细胞分化和血管的发生形成通路中存在其他基因的影响,需要做进一步的研究。
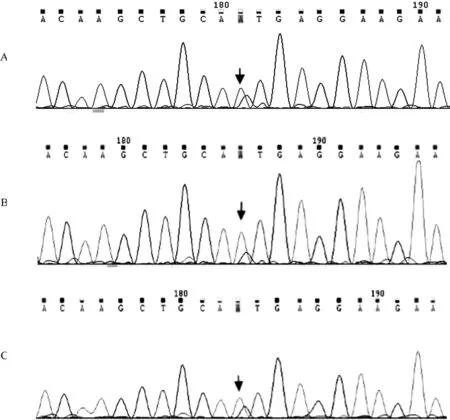
A为患儿AGGF1基因第6外显子序列;B为患儿父亲AGGF1基因第6外显子
综上所述,KTS是一类以血管发育异常为主的少见疾病,其病因与发病机制仍在不断研究探索中,若小儿时期能够早期发现,明确病变类型和程度,可尽早施行正确、有效的个体化治疗方案。
[1]刘建平,刘风恩,段训洪,等.静脉畸形骨肥大综合征[J].中国小儿血液与肿瘤杂志,2015,12(20):284-285.
[2]SIM N L,KUMAR P,HU J.SIFT web server:predicting effects of amino acid substitutions on proteins[J].Nucleic Acids Res,2012,40 (Web Server issue):W452-W457.
[3]ADZHUBEI I A,SCHMIDT S,PESHKIN L,et al.A method and server for predicting damaging missense mutations[J].Nat Methods,2010,7(4):248-249.
[4]SCHWARZ J M,COOPER D N,SCHUELKE M,et al.Mutation taster 2:mutation prediction for the deep-sequencing age[J].Nat Methods,2014,4(11):361-362.
[5]HUSMANN D A,RATHBURN S R,DRISCOLL D J.Klippel-Trenaunay syndrome:incidence and treatment of genitourinary sequelae[J].J Urol,2007,177(4):1 244-1 249.
[6]SUNG H M,CHUNG H Y,LEE S J,et al.Clinical experience of the klippel-trenaunay syndrome[J].Arch Plast Surg,2015,42(5):552-558.
[7]徐霖,杨守俊,陈平有,等.Klippel Trenaunay综合征的造影检查与鉴别诊断[J].郧阳医学院学报,2002,21(2):111-112.
[8]PALLER A S,MANCINI A J.Vascular disorders of infancy and childhood[M]// Hurwitz clinical pediatric dermatology:a textbook of skin disorders of childhood and adolescence.3rd.Philadelphia,PA:Elsevier Saunders,2006:307-344.
(本文编辑:张红)
Clinical and genetic analysis of a child with klippel-trenaunay syndrome.
ZHAO Lijun,TIAN Yongli,YANG Yuehong,KOU min
( Shanxi Children′s Hospital,Tianyuan 030013,China)
Objective:To investigate the clinical manifestations and molecular basis of a child with Klippel-Trenaunay syndrome(KTS).Methods:Clinical features,and laboratory data were collected.Genomic DNA was extracted from leukocytes of peripheral blood of the patient,detected by polymerase chain reaction,and the mutations identified by direct sequencing.Results:KTS was diagnosed on the basis of comprehensive consideration of clinical presentations,and laboratory test results.This gene mutation test revealed a nucleotide substitution of guanine for adenine at the position 923 of exon 6 of AGGF1 gene (c.923A>G),which caused a missense mutation of asparagine to serine at codon 923 (p.Asn923Ser).Loci conservation analysis and functional prediction of missense mutation of AGGF1 protein revealed that c.923A>G was a pathogenic mutation.Conclusion:One case of KTS was diagnosed by clinical and laboratory examinations.From the aspect of molecular genetics,AGGF1 gene mustation has confirmed the diagnosis of KTS in this patient.
klippel-trenaunay syndrome;gene mutation;child
王利荣(1965— ),女,山西省五台县人,学士学位,副主任医师,主要从事心内科工作。
1671-8631(2017)05-0354-05
R394
B
2016-12-27
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中华实用诊断与治疗杂志(2022年1期)2022-08-31
现代仪器与医疗(2022年1期)2022-04-19
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年2期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国医药指南(2017年3期)2017-11-13
湖北农业科学(2014年11期)2014-09-10
