
利用液相色谱–质谱联用技术研究二元表面活性剂体系–啶虫脒微乳剂热贮稳定性*
2017-06-05 14:14赵振东谢艳丽
化学分析计量 2017年3期
赵振东,谢艳丽
利用液相色谱–质谱联用技术研究二元表面活性剂体系–啶虫脒微乳剂热贮稳定性*
赵振东1,2,谢艳丽2
(1.海南国际旅游岛发展研究院,海口 570228; 2.海南大学材料与化工学院,海口 570228)
应用液相色谱–质谱联用技术评价复配的阴/非离子二元表面活性剂复合体系–啶虫脒微乳剂的热贮稳定性。将二元表面活性剂复合体系–啶虫脒微乳剂于(54±2)℃下贮藏14天,取出后于室温下干燥24 h,用液相色谱–质谱联用仪测定啶虫脒含量。该法线性拟合方程为y=97 060.72x+542 587.4,线性范围为0.01~0.25 mg/L,相关系数为0.999 8。方法检出限为0.001 mg/L(S/N=3),色谱保留时间和色谱峰面积重复性分别为0.31%,1.44%(n=6),加标回收率为86.23%~107.20%。该法具有简单、准确和灵敏的特点。
液相色谱–质谱联用技术;表面活性剂;啶虫脒;热贮稳定性
农药乳油使用大量有毒溶剂,严重污染环境。农药微乳剂以水替代有机溶剂,是当今研究热点之一。微乳液是由油、水、表面活性剂组成的有序多元体系,具有界面面积较大、界面张力超低、热力学稳定等性质。由于其特殊组成及结构,微乳液作为有机分子聚集体,具有很强的增溶、增敏及增稳作用[1–2]。O/W型微乳液作为药物载体已广泛应用于农药等领域。
啶虫脒是高效广谱内吸性新型杀虫剂,能够有效防治果蔬蚜虫,同时能够用来防治对有机磷、氨基甲酸酯以及拟除虫菊酯类等产生耐药性的害虫[3–5],它具有低毒,环境污染小的特点。其中3%啶虫脒微乳剂已经得到了广泛应用。
当前微乳剂多采用单一性质的表面活性剂作为助剂。在实际应用中,由二元体系表面活性剂混合物形成的胶束聚集体常显示出与单一表面活性剂明显不同的特性,还具有优越的协同增效作用,对实现高效率的微乳液性能十分重要。热贮稳定性关系到药品贮运、降解和药效,笔者拟采用阴/非离子二元表面活性剂混合体系复配啶虫脒微乳液,并考察其热贮稳定性指标。
啶虫脒的分析方法主要有气相色谱法、气相色谱–质谱联用法、液相色谱法和液相色谱–质谱联用法(LC–MS)或液相色谱–串联质谱法(LC–MS/MS)[6–18]。其中气相色谱法和气相色谱–质谱联用法主要用于分析挥发性组分。水基啶虫脒微乳液必须经过复杂的前处理,有效提取啶虫脒组分,除去难挥发水溶性基质方可用气相色谱法检测,分析过程繁琐,成本高。相比较而言,液相色谱法不需要进行样品前处理,特别适用于水溶性微乳剂类样品分析。采用液相色谱–紫外检测器即可直接测定啶虫脒含量,该法缺点是灵敏度偏低。啶虫脒属氨基甲酸酯类化合物,可以采用液相色谱–柱后衍生–荧光检测的方法,有效提高响应灵敏度,但实验过程略显繁琐,衍生化试剂成本高,毒性大,易造成环境二次污染。液相色谱–质谱联用技术已经非常成熟,该法结合液相色谱强分离能力和质谱的高灵敏度特点,普适性和专一性都非常强,特别适用于水基化微乳剂的测定,而且无需样品前处理。
笔者拟采用LC–MS技术直接测定阴/非离子二元表面活性剂复合体系–3%啶虫脒微乳剂中啶虫脒的含量,并据此评价该微乳剂的热贮稳定性。
1 实验部分
1.1 主要仪器与试剂
超高效液相色谱–高分辨质谱联用仪:LC–MS–IT–TOF型,日本岛津公司;
数显恒温水浴锅:HH–6型,常州澳华仪器有限公司;
电子天平:AUY220型,日本岛津公司;
磁力加热搅拌器:DF–101S型,金坛市医疗仪器厂;
超纯水系统:Purelab Ultra型,美国Pall公司;
乙腈:HPLC级,美国Fisher公司;
啶虫脒:纯度为99%,德国Dr. Ehrenstofer GmbH公司;
乳化剂:由阴离子表面活性剂和多种非离子表面活性剂复配而成;
实验用水均由超纯水系统制备,电阻率为18.2 MΩ·cm。
1.2 实验方法
1.2.1 微乳剂制备
将定量溶剂及原药加入反应器中,搅拌至完全溶解。再加入阴离子表面活性剂及多种非离子表面活性剂等助剂,用水定容,充分搅拌即得到3%啶虫脒阴/非离子二元表面活性剂复合体系微乳剂。
1.2.2 热贮稳定性考察
称取一定量微乳剂,密封于广口瓶中,置于恒温箱中于(54±2)℃贮藏14天,取出,放入干燥器中,室温冷却后于24 h内测定啶虫脒含量。
1.2.3 实验步骤
样品用乙腈稀释,经0.45μm有机膜过滤上机分析,每样平行测定3次。
1.3 仪器工作条件
1.3.1 液相色谱条件
色谱柱:Acquity HSS–T3柱(100 mm×2.1 mm,
1.8μm,美国Waters公司);柱温:45℃;进样体积:2 μL;流动相:1‰甲酸水溶液–乙腈,流量为0.3 mL/min,梯度洗脱程序见表1。

表1 梯度淋洗程序
1.3.2 质谱条件
ESI正负离子同时扫描模式;正离子电压:4.5 kV;负离子电压:–3.5 kV;雾化气:氮气,流量为1.5 L/min;干燥气:氮气,压力为100 kPa;冷却气:氩气;检测器电压:1.8 kV;Heat Block 温度:200℃;CDL 温度:200℃;扫描范围:m/z200~300;离子累积时间:15 ms。
2 结果与讨论
2.1 质谱参数的优化
正负离子同时扫描模式灵敏度优于单一正离子扫描模式。单一扫描模式下,相反电荷离子占据喷雾针表面,中和部分电荷,减少了目标离子数量,从而降低了灵敏度。而正负离子同时扫描时,电场力推开喷雾针表面过剩相反电荷,从而提高响应灵敏度。故选择正负离子同时扫描模式。
此外,质荷比范围越窄,响应灵敏度越高,这是背景离子减少、目标离子数量增加所致。离子累积时间直接关系离子数量,累积时间过短则灵敏度低,累积时间过长则影响峰形及采集曲线点数。综合考虑,选择离子累积时间为15 ms。
2.2 色谱柱选择
比较两根常用沃特世超高效色谱柱(HSS–T3 和C18)对啶虫脒的分离效果。结果发现两根色谱柱并无明显区别,最终选用HSS–T3色谱柱。
2.3 流动相选择
试验比较甲醇–水和乙腈–水对啶虫脒的分离效果,二者差异不明显。由于乙腈对一些弱极性杂质清除能力更强,最终选择乙腈–水作为流动相。啶虫脒在流动相中主要以[M+H]+(m/z223.074 5)离子形式存在。若添加少量有机酸,可以有效增加液相中[M+H]+离子数量,从而提高灵敏度。试验了不同比例甲酸的流动相,结果表明随着甲酸含量增加,啶虫脒响应强度明显提高;当甲酸含量达到1‰时灵敏度基本稳定,再增加无明显效果。最终选择1‰甲酸水溶液–乙腈体系作为流动相。梯度洗脱的目的是为了有效分离啶虫脒与表面活性剂,从而保证检测灵敏度。
2.4 基质效应
电喷雾质谱存在较强基质效应,原因主要是不同基质增强或抑制目标物气相离子的形成效率。微乳剂样品中含有大量的阴离子和非离子表面活性剂,它们具有良好的疏水和亲水两性性质。一方面,在喷雾液滴形成过程中,更容易占据液滴表面,阻碍目标物向液滴表面转移,从而抑制目标物气相离子的形成。另一方面,带有相反电荷的表面活性剂中和了部分目标物离子,降低了液滴电荷密度,不利于形成更小液滴。以上原因均导致灵敏度下降。
为减少基质效应影响,一方面通过分离方法优化,减少啶虫咪出峰时杂质的干扰。另一方面,通过试验比较空白基质加标和溶剂响应值变化,结果发现依然存在明显的抑制效应。因此,最终采用空白基质配制系列标准溶液绘制工作曲线,以保证标准溶液与样品响应一致,提高测量结果的准确度。
2.5 离子流色谱图
在1.3仪器工作条件下,将二元表面活性剂体系–啶虫脒微乳剂样品用乙腈逐级稀释后直接进样分析,离子流色谱如图1所示。由图1可见,啶虫脒与样品基质色谱峰分离良好,色谱峰形尖锐对称,便于定量。
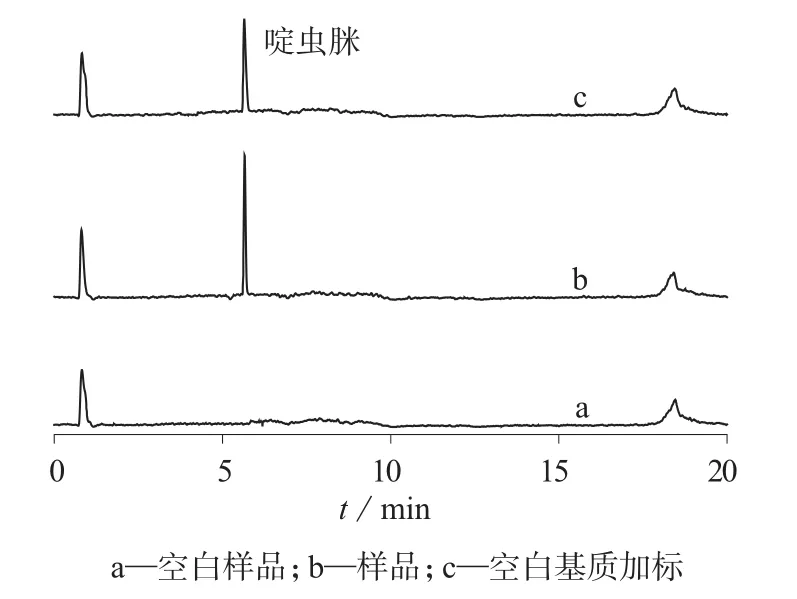
图1 二元表面活性剂体系–啶虫脒微乳剂的离子流色谱图
2.6 线性方程与线性范围
精密称取一定量啶虫脒对照品于10 mL容量瓶中,用甲醇溶解并定容,得到1 000 mg/L啶虫脒标准储备液。按照1.2实验方法制备空白基质,然后取一定体积储备液用空白基质逐级稀释成0.01,0.025,0.05,0.1,0.25 mg/L系列标准工作溶液。
在1.3.1色谱条件下依次分析系列标准溶液,啶虫脒的保留时间为5.67 min。以标准工作溶液的质量浓度(x,mg/L)为横坐标,色谱峰面积响应值(y)为纵坐标,拟合得线性方程y=97 060.72x+542 587.4,线性范围为0.01~0.25 mg/L,相关系数(r)为0.999 8。
2.7 检出限和定量限
采用逐级稀释方法,截取一段噪音信号,利用3倍信噪比作为检出限,10倍信噪比作为定量下限,计算得其检出限为0.001 mg/L,定量下限为0.003 mg/L。
2.3 两组血脂及CRP水平比较 治疗后,两组血脂指标比较,差异均无统计学意义(P>0.05)。治疗前,A组与B组CRP水平比较[(4.84±1.29)mg/L比(4.82±1.34)mg/L],差异无统计学意义(P>0.05);治疗后,两组CRP水平均降低,且B组为(3.47±0.76)mg/L,低于A组的(3.79±0.65)mg/L,差异有统计学意义(P<0.05)。
2.8 精密度试验
按照1.2.1的方法制备0.02 mg/L啶虫脒基质标准溶液,连续测试6针,记录每针的色谱保留时间和色谱峰面积,计算相对标准偏差,相关数据见表2。由表2可知,色谱保留时间及色谱峰面积6次测量结果的相对标准偏差分别为0.31%和1.44%。
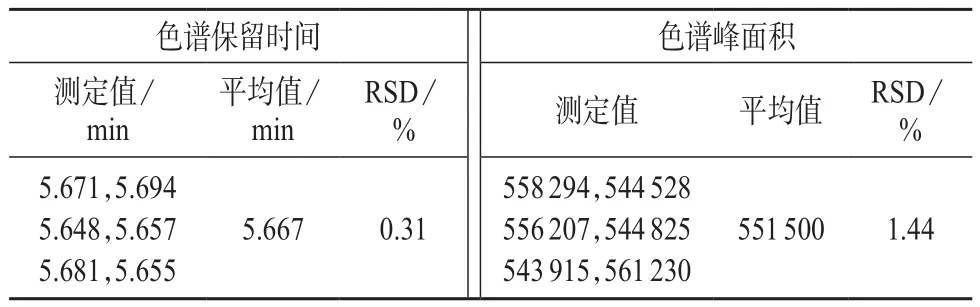
表2 精密度试验结果
2.9 加标回收试验
按照1.2.1方法制备空白基质加标溶液,逐级稀释后分别添加啶虫脒0.05,0.1,0.5 mg/L。样品经0.45 μm有机膜过滤后直接进样分析,加标回收试验结果见表3。
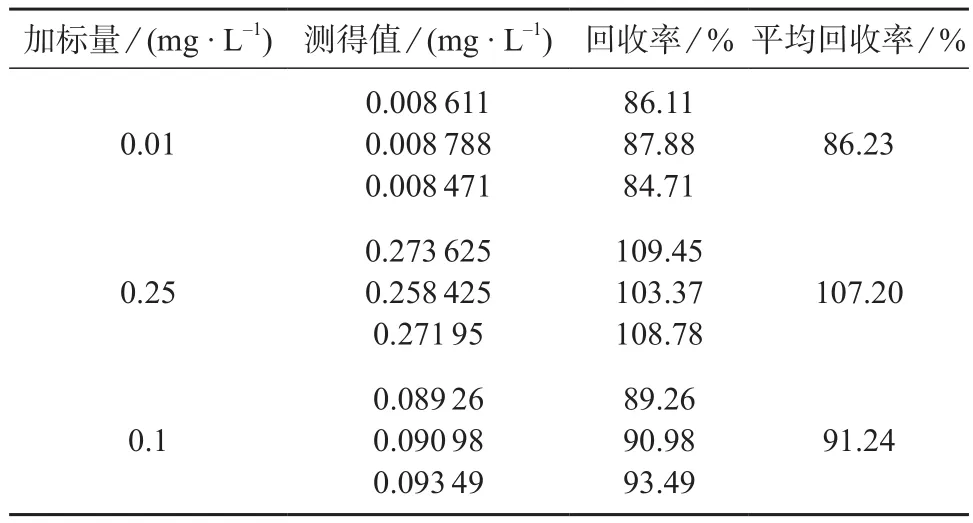
表3 加标回收试验结果
由表3可知,3个加标水平回收率为86.23%~107.20%,表明本法测量准确度较高。
2.10 样品测定
在1.3.1色谱条件下,将样品用乙腈逐级稀释后直接进样分析(如图1所示),每个样品平行测定3次,结果见表4。由表4可知,二元表面活性剂复合体系–3%啶虫脒乳剂配方于(54±2)℃恒温14天后分解率<5%,符合热贮稳定性要求。
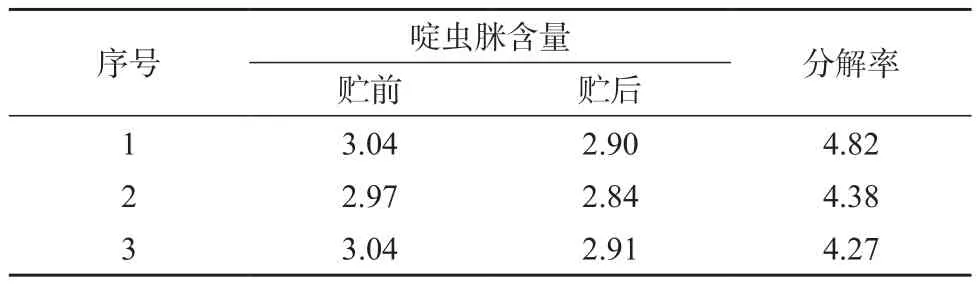
表4 二元表面活性剂复合体系–3%啶虫脒乳剂热贮稳定性试验结果 %
3 结语
采用LC–MS技术直接测定阴/非离子二元表面活性剂复合体系–3%啶虫脒微乳剂含量,该法具有操作简单、准确和灵敏度高等特点。
[1]Garti N, Yaghmur A, Aserin A, et al. Solubilization of active molecules in microemulsions for improved environmental protection [J]. Colloids and Surfaces A: Physicochem Eng Aspects, 2004,230: 183–190
[2]安红丽.非离子型表面活性剂微乳的研制[J].西北农林科技大学学报,2007,35(3): 65–69.
[3]齐崇广,任元宾,李艳芬,等. 3%啶虫脒微乳剂的稳定性研究[J].应用化工,2003,32(4): 54–56.
[4]钱长英. 3%啶虫脒微乳剂的研究[J].安徽化工,2003(1): 36–37.
[5]王凤芝,李波,齐慧芹,等. 5%啶虫脒微乳剂的研制[J].农药科学与管理,2006,25(9): 33–36.
[6]孙楠,薛健.气相色谱法测定金银花中残留的啶虫脒[J].农药,2007,46(4): 256–257.
[7]李晔,郭蒙京,袁佗.气相色谱–质谱法测定茶叶中溴虫腈、啶虫脒、茚虫威、三氯杀螨醇及拟除虫菊酯等11种农药残留[J].中国卫生检验杂志,2016(11): 1 579–1 581.
[8]侯宏安,张卫兵,苏国宝.分散固相萃取– GC/MS法测定人血中啶虫脒的含量[J].刑事技术,2012 (2): 30–31.
[9]梁林,潘金菊,刘伟.气相色谱–负化学离子源–质谱法测定黄瓜中啶虫脒残留[J].农药科学与管理,2012,33(9): 25–28.
[10]毕富春.啶虫脒高效液相色谱法定量分析[J].现代农药,2002 (3): 16–17.
[11]王素茹,陈丽霞,林靖凌.液相色谱法测定甘蓝中啶虫脒残留[J].热带农业科学,2012,10(30): 27–29.
[12]许鹏军,张红艳,陶晡,等.高效液相色谱法测定黄瓜和油菜中的啶虫脒残留量[J].分析试验室,2008,27(10): 80–83.
[13]王骏,钟青,郭忠成,等.烟草中啶虫脒、吡虫啉残留量的UPLC–MS/MS测定[J].烟草化学,2010 (10): 41–44.
[14]吴星祥,杨娟,李盛安,等.液相色谱–串联质谱法测定番茄中多菌灵和啶虫脒的残留量[J].现代农业科技,2012(5): 196–198.
[15]张海超,艾连峰,郭春海,等.液相色谱–串联质谱法测定水果蔬菜中多菌灵、吡虫啉、啶虫脒与甲基硫菌灵的残留量[J].分析测试学报,2014,33(3): 295–300.
[16]Geetha G,Sreenivas C,Shashi V. Determination of pesticide residues in spinach[J]. Indian J Appl Res,2016,6(11): 395–397.
[17]Aline B,Luciane M,Lademir L,et al. Determination of pesticide residues in whole wheat flour using modified QuEChERS and LC–MS/MS[J]. Food Anal Methods,2017(10): 1–9.
[18]Zhong Q,Shen L,Liu J,et al. Pre-column dilution large volume injection ultra-high performanceliquid chromatography-tandem mass spectrometry for the analysis of multi-class pesticides in cabbages[J]. J Chromatogr A,2016,1 442: 53–61.
一种测定木制品中木馏油的方法
申请(专利权)人:通标标准技术服务(上海)有限公司
摘要本发明涉及一种测定木制品中木馏油的方法,包括以下步骤:(1)称取粉碎且混合均匀的试样,加入提取溶剂甲苯、示踪剂和内标标准溶液,涡旋混合均匀,超声提取;(2)采用气相色谱质谱联用仪,对所得样品提取溶液中的木馏油标记物进行测定;(3)根据步骤(2)所得的色谱图和质谱图,分析确认样品提取溶液中是否含有木馏油特有的多环芳烃标记物或酚类标记物,以判定样品中是否含有相应的木馏油;再对被选作木馏油标记物的多环芳烃和酚类化合物进行鉴别和定量;最终,由样品中标记物的含量经换算得到样品中木馏油的含量;本发明方法快速、简单,通过半定量的方法对法规所禁止使用的4种木馏油在样品中的含量进行了测定,检测限均为50 mg/kg。
一种瘦肉精类检测方法
公开(公告)号:CN106546545A 公开(公告)日:2017.03.29
申请(专利权)人:无锡艾科瑞思产品设计与研究有限公司
摘要本发明公开了一种瘦肉精类检测方法,取5~12 g待测样品,绞碎至粒径0.18~0.28 mm(60~80目),加入15~30 mL高氯酸溶液,在70~80℃的温度下超声提取20~30 min,冷却至室温,离心,取上清液,沉淀加4~8 mL高氯酸溶液洗涤,离心取上清液,合并两次上清液,用氢氧化钠溶液调节pH,先用20~30 mL乙醚,振荡提取20 min,回收有机相,再用15~25 mL乙醚萃取两次,合并有机相蒸干,用盐酸溶液溶解蒸干固体,用去离子水定容至10 mL,得到待测样品;采用紫外可见分光光度计测定吸光度,由标准曲线得到待测样品中瘦肉精类的含量。本发明测定结果准确度高;操作简单、测定成本低;检测反应速度快,使用方便,产生的废液几乎不产生二次污染,检测范围宽、最低检出限低、灵敏度高。
Study on Stability of Thermal Storage in Binary Ionic/Nonionic Surfactants–Acetamiprid Microemulsion by LC–MS
Zhao Zhendong1, 2, Xie Yanli2
(1. Institute for the Development of Hainan International Tourism Island, Haikou 570228, China; 2. College of Materials and Chemical Engineering, Hainan University, Haikou 570228, China)
Study on thermal storage stability of binary ionic/nonionic surfactants-acetamiprid microemulsion was developed by LC–MS. After being stored for 14 day at (54±2)℃, the binary ionic/nonionic surfactants-acetamiprid microemulsion was dried for 24 h at room temperature, and acetamiprid in it was determined by LC–MS. The results showed good linearity with linear equation ofy=97 060.72x+542 587.4, ranging from 0.01 mg/L to 0.25 mg/L, the correlation coefficient was 0.999 8. The detection limit was 0.001 mg/L. The repeatability of chromatograph retention time and peak area was 0.31% and 1.44%(n=6), respectively. The standard recoveries were between 86.23% and 107.20%. The method is simple, reliable and sensitive.
LC–MS; surfactant; acetamiprid; stability of thermal storage
O657.7
A
1008–6145(2017)03–0027–04
*海南省自然科学基金项目(20152020);国家重大科学仪器设备开发专项(2012YQ090229)
联系人:赵振东;E-mail: zhaozhd898@126.com
2017–02–18
10.3969/j.issn.1008–6145.2017.03.006
猜你喜欢
新型工业化(2022年3期)2022-06-18
当代水产(2022年4期)2022-06-05
口腔护理用品工业(2021年4期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
油气田环境保护(2015年4期)2015-12-28
中国造纸(2015年7期)2015-12-16
中国洗涤用品工业(2015年2期)2015-02-28
中国洗涤用品工业(2015年2期)2015-02-28
天然气与石油(2015年2期)2015-02-28
