
气相色谱–串联质谱法快速测定茶叶中的蒽醌*
2017-06-05 14:14何正和魏斌魏云计朱臻怡何健冯民张科秦娴
化学分析计量 2017年3期
何正和,魏斌,魏云计,朱臻怡,何健,冯民,张科,秦娴
[淮安出入境检验检疫局,国家饲料安全检测重点实验室(淮安),江苏淮安 223001]
气相色谱–串联质谱法快速测定茶叶中的蒽醌*
何正和,魏斌,魏云计,朱臻怡,何健,冯民,张科,秦娴
[淮安出入境检验检疫局,国家饲料安全检测重点实验室(淮安),江苏淮安 223001]
建立气相色谱–串联质谱法(GC–MS–MS)快速测定茶叶中蒽醌的方法。样品中的蒽醌用色谱纯乙腈提取,经过涡旋1 min,超声20 min,离心、浓缩后用固相萃取柱(SPE)净化,以DB–5MS毛细管柱分离,以m/z207.9,152为定性离子对、m/z207.9,180为定量离子对,多反应检测模式下测定,外标法定量。蒽醌的质量浓度在1~500 μg/L范围内与定量离子的峰面积呈良好的线性关系,相关系数为0.999 98,方法的检出限为1 μg/kg。在10,20,50 μg/kg加标水平下,样品加标回收率为92.85%~98.10%,测定结果的相对标准偏差为8.50%~11.05%(n=5)。该方法简单、快速,检出限低,准确度和精密度高,可用于茶叶中蒽醌的检测。
茶叶;蒽醌;固相萃取;气相色谱–串联质谱法
茶叶在中国由来已久,随着经济全球化,茶叶开始流向国外,出口量不断增多,国外对茶叶品质的要求也越来越苛刻。蒽醌(Anthraquinone,CAS#84–65–1)为茶叶中的有害物质之一,2014年10月,欧盟发布的(EU)No1146/2014要求茶叶中蒽醌的最高限量为0.02 mg/kg[1]。近两年茶叶出口中,由于蒽醌超标就被通报数十次[2],给我国茶叶出口市场带来巨大的冲击。导致这一后果有多方面的因素,其中最主要的是茶叶前处理基质较为复杂,国内还没有针对茶叶中蒽醌检测的标准[3–5],定量测定蒽醌的文献也不多。为了保证国内茶叶产品的质量安全及茶叶出口的“丝绸之路”,建立茶叶产品中蒽醌残留量快速、准确的检测方法十分必要。目前,蒽醌类定量测定的方法主要有分光光度法[6–7]、高效液相色谱法[8–9]、胶束电动毛细管色谱法[10]、气相色谱–质谱法[11]、气相色谱–串联质谱法[12]及比色定量法[13–14]等,其中多数方法测定的是蒽醌类化合物的总量。气相色谱–串联质谱法由于定性可靠,检测灵敏度高,抗干扰能力强等优点,成为化学污染物多残留检测的研究热点[15–16]。2015年,柴芸彬利用GPC净化处理茶叶中复杂基质,能够较好地纯化分离出蒽醌,采用气相色谱–串联质谱法定量[12],但此方法需凝胶色谱净化系统,测定过程较为耗时。
笔者针对茶叶前处理中的复杂基质,采用弗罗里硅土SPE小柱净化,用气相色谱串联质谱法(GC–MS–MS)选择离子扫描测定蒽醌的含量。该方法可以很好地分离出蒽醌并进行定量测定,较其它方法快速高效。同时针对蒽醌不溶于水,易溶于有机溶剂这一特性,利用乙腈、乙醇、乙酸乙酯等有机溶剂作为提取剂来探讨蒽醌的提取效率,进而建立一个能够满足茶叶蒽醌检测要求的快速方法。该方法具有样品前处理简单、快速,检出限低,准确度和精密度高等优点。
1 实验部分
1.1 主要仪器与试剂
气相色谱质谱联用仪:Angilent 7890B–7000C型,美国安捷伦科技有限公司;
九阳料理粉碎机:C012型,九阳股份有限公司;
电子天平:PB–203E型,瑞士梅特勒–托利多公司;
超声仪:SB–5200型,广州沪瑞明仪器有限公司;
漩涡混合器:WH–86型,江苏盛蓝仪器制造有限公司;
离心机:3K15型,德国Sigma公司;
氮吹仪:N–EVAP14165型,美国Organomation公司;
SPE小柱:1 000 mg/6 mL,弗罗里硅土,河北津扬滤材厂;
针式尼龙滤膜:0.22 μm,天津亳津科技有限公司;
乙腈、丙酮:HPLC级,美国Honeywell公司;
乙腈:分析纯,南京化学试剂有限公司;
三氯甲烷:HPLC级,美国VBS公司;
正己烷、甲醇、乙醇:HPLC级,德国Merck公司;
乙酸乙酯、乙醚、氯化钠、硫酸钠:分析纯,南京化学试剂有限公司;
蒽醌(Anthraquinone,CAS#84–65–1)标准品:纯度为99.0%,德国Dr Ehrenstorfer公司;
实验室用水为去离子水。
1.2 标准溶液的配制
准确称量30.9 mg的蒽醌标准品于10 mL的容量瓶中,用正己烷定容至标线,然后稀释成10 μg/mL的蒽醌标准储备液,于4℃冰箱中避光保存。
吸取一定体积的蒽醌标准储备液,用基质空白溶液逐级稀释成蒽醌的质量浓度分别为1,5,10, 50,100,500 μg/L系列标准工作溶液。
1.3 样品处理
将茶叶用粉碎机粉碎,过0.83 mm(20目)筛,混匀,常温存放在100 mL干净的棕色玻璃瓶中。精确称取2.0 g(精确至0.01 g)粉碎后的茶叶样品至50 mL离心管中,加入10 mL去离子水,涡旋30 s,放入45℃水浴锅超声20 min,冷却至室温后加入8 mL色谱纯乙腈,涡旋1 min。离心管中加入1~3 g氯化钠恒重,在离心机中于10 000 r/min离心3 min。将上清液转移至试管中,用6 mL色谱纯乙腈再提取一次,合并有机相,氮吹至约2 mL。SPE小柱加2 cm高度的无水Na2SO4,用3 mL正己烷–丙酮溶液(体积比为8∶2,下同)活化,将氮吹后的浓缩液转移至SPE小柱,再用2 mL正己烷–丙酮溶液洗涤氮吹管2~3次,并将洗涤液移入SPE小柱中,用8 mL正己烷–丙酮溶液洗脱,滤液用氮气吹至尽干,用丙酮定容至1 mL,过0.22 μm针式尼龙滤膜于色谱进样瓶中,用气相色谱–串联质谱仪测定。
1.4 仪器工作条件
1.4.1 色谱条件
色 谱 柱:Agilent122–5532UI DB–5MS柱(30 m×0.25 mm,0.25 μm,美国安捷伦科技有限公司);进样口温度:280℃;载气:高纯氦,流量为1.0 mL /min;不分流进样;进样量:1 μL;柱温箱升温程序:初始温度70℃,保持1 min,以10℃/min升至280℃,保持2 min。
1.4.2 质谱条件
电离模式:电子轰击(EI),70 eV;扫描方式:多反应检测模式(MRM);离子源温度:230℃;循环周期:24 s;色谱过滤峰宽:0.8 s;碰撞气:高纯氮气,流量为1.5 mL/min;溶剂延迟:5 min;母离子、子离子、碰撞能量及驻留时间参数见表1。
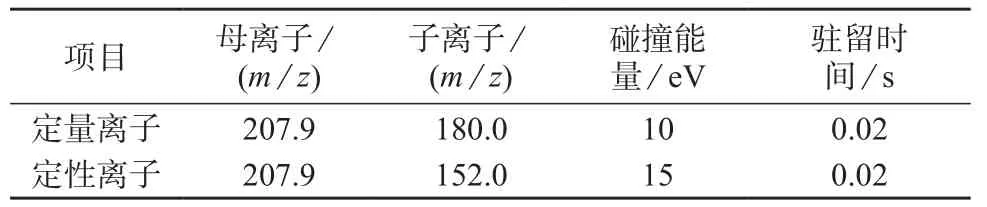
表1 多反应监测模式(MRM)参数
1.5 色谱图与质谱图
在1.4仪器工作条件下,对100 μg/L的蒽醌标准工作溶液、20 μg/kg空白白茶加标样品进行分析,MRM离子流色谱图分别见图1、图2。
2 结果与讨论
2.1 气相色谱–质谱条件的选择
对于茶叶中蒽醌污染物,笔者采用一般农残分离的DB–5MS石英毛细管柱(30 m×0.25 mm,0.25 μm);载气流速为1.0 mL/min。通过不断调整进样口温度、色谱柱温控程序、载气流速的色谱关键参数,避免基质干扰峰与目标化合物保留时间的重叠。在尽量提高分离度的情况下,缩短分析时间。最后经过优化得到合适的色谱柱升温条件:70℃保持1 min,然后以10℃/min程序升至280℃,保持2 min。在优化的色谱条件下,采用Q3 SCAN全扫描方式得到蒽醌化合物质谱图,选择2~3个相对丰度较高、质核比较大的作为产物离子。再将质谱的方法改为产物离子扫描方式对选定的产物离子进行扫描,分别选择丰度较大的两组离子m/z207.9,152,m/z207.9,180为定性及定量离子对。为了保证检测的灵敏度和准确性,选用不同大小的碰撞能量观察两组离子的响应情况,结果发现定性及定量离子对合适的碰撞能量分别为15,10 eV。得到的蒽醌化合物MRM离子参数见表1。
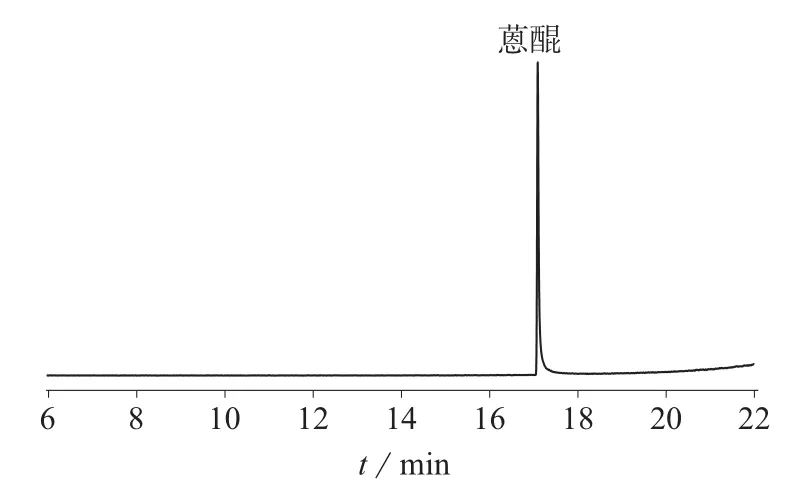
图1 100 μg/L的蒽醌标准品MRM离子流色谱图

图2 空白白茶加标样品MRM离子流色谱图
2.2 样品前处理方式的选择
将具有代表性茶叶样品存放在500 mL干净的玻璃瓶中。将茶叶用粉碎机粉碎,过0.83 mm(20目)筛,混匀,常温存放在100 mL干净的棕色玻璃瓶中。
针对茶叶样品的复杂基质,为了提高提取效率,笔者将样品加入10 mL 去离子水涡旋并在45℃水浴锅超声,再用色谱纯乙腈提取两次。在纯化过程中,选择N-丙基乙二胺(PSA)、石墨碳黑(GCB)和弗罗里硅土SPE小柱进行净化,发现前两者较后者易吸附目标化合物且净化不够充分,试验结果见表2。对于前两者净化效果不好的原因文献中也有报道[17],GCB会吸附平面型结构的化合物。因此采用弗罗里硅土SPE小柱纯化分析样品。

表2 不同萃取柱的空白添加样品测定值
2.3 萃取溶剂的选择
按1.3进行操作,对提取溶剂进行筛选试验,通过改变不同的提取溶剂测定蒽醌的含量。其中丙酮、甲醇和乙醇除不加水外处理步骤如1.3。不同溶剂的加标回收率见表3。
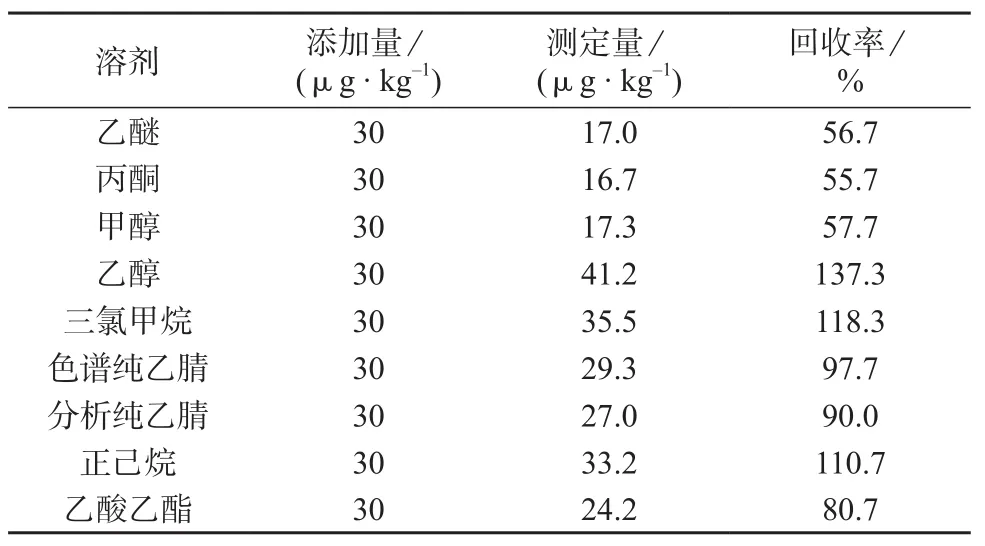
表3 不同溶剂对样品的加标回收率测试结果
由表3可以看出,提取效率最好的是色谱纯乙腈;乙醚、丙酮和甲醇的提取效率较低,并且丙酮和甲醇由于提取过程中没有加水,直接提取后的基质较为复杂;三氯甲烷和乙醇的提取结果偏高,乙醇提取同样也存在基质较为复杂问题;分析纯乙腈和正己烷的提取效果较色谱纯乙腈差,同时正己烷提取的稳定性不好。因此选择色谱纯乙腈作为提取溶剂。
2.4 线性方程、线性相关系数与检出限
为了避免基质对蒽醌检测的影响,采用基质标准溶液建立标准工作曲线。按1.4仪器工作条件测定1.2配制的系列基质标准工作溶液,以蒽醌的质量浓度(X)为横坐标、以定量离子的峰面积(Y)为纵坐标绘制标准工作曲线,计算得线性回归方程为Y=965.16X–217.85,线性范围为1~500 μg/L,相关系数r=99.998%。以信噪比S/N=3确定方法的检出限,信噪比S/N=10确定方法定量限,检出限、定量限分别为1,5 μg/kg。
2.5 精密度试验
在1.4仪器工作条件下,对添加10,20,50μg/kg 3个浓度水平的空白茶叶样品进行测定,结果见表4。
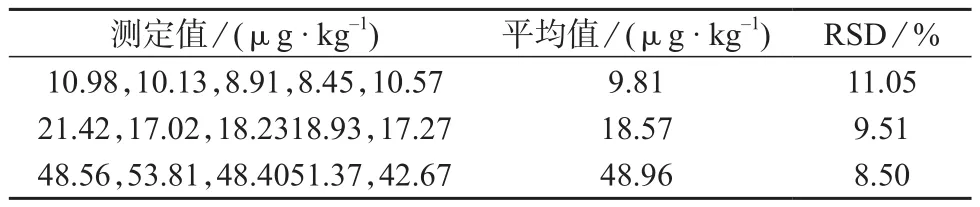
表4 精密度试验结果
由表4可知,测定结果的相对标准偏差为8.50%~11.05%,表明该方法的精密度较好,可以满足茶叶中蒽醌检测要求。
2.6 回收试验
准确称量2.0 g(精确至0.01 g)不含检测目标化合物的空白茶叶样品,在空白茶叶样品中分别添加10,20,50 μg/kg的蒽醌,按1.3进行样品处理,在1.4仪器工作条件下测定,进行加标回收试验,结果见表5。
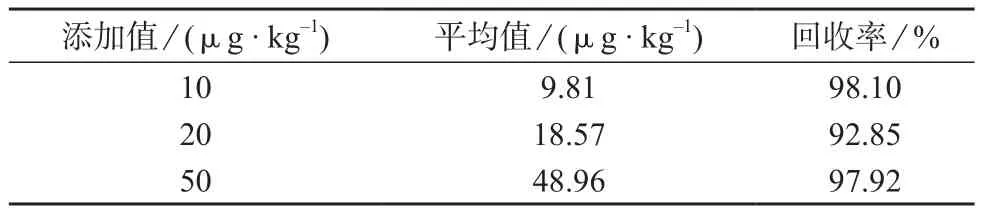
表5 加标回收试验结果
由表5可知,3种浓度的空白样品加标回收率在92.85%~98.10%之间,表明该方法具有较高的准确度,可以满足茶叶中蒽醌检测要求。
2.7 样品测试
按照1.3用色谱纯乙腈为提取溶剂对市场购买的不同品种茶叶进行样品处理,在1.4仪器工作条件下平行测定3次,测定结果平均值见表6。由表6结果可知,竹叶青和铁观音的蒽醌含量低于欧盟(EU)No 1146/2014 规定的茶叶中蒽醌最大残留限量(MRL)0.02 mg/kg;而龙井、毛峰和白茶中蒽醌的含量高于此值。

表6 样品的测试结果
3 结语
以色谱纯乙腈为萃取剂,弗罗里硅土SPE小柱净化等前处理,经气相色谱–串联质谱法分析,建立了茶叶中蒽醌残留的检测方法。该方法快速高效,检出限低,准确度和精密度高,能够满足欧盟茶叶中蒽醌限量要求(<0.02 mg/kg),可用于茶叶蒽醌的检测。通过几类茶叶样品的测试发现蒽醌存在于茶叶中是很普遍的现象。目前定量测定的方法不多,国标中蒽醌的分析也只停留在定性阶段。为了能够给国内茶叶出口提供技术支持,茶叶中蒽醌的来源分析也是以后研究的方向。只有从源头上控制其含量,才能进一步促进我国的茶叶贸易。
[1]COMMISSION REGULATION (EU)No 1146/2014 of 23 October 2014. Amending Annexes II, III, IV and V to Regulation (EC) No 396/2005 of the European Parliament and of the Council as regards maximum residue levels for anthraquinone, benfluralin, bentazone, bromoxynil, chlorothalonil, famoxadone, imazamox, methyl bromide, propanil and sulphuric acid in or on certain products [R]. European Union: Official Journal of the European Union,2014,L308: 3–59.
[2]朱凤玲. 2014年欧美日通报我国不合格茶叶信息汇总[J].中国茶叶,2015(2): 27–28.
[3]GB/T 23204–2008 茶叶中519种农药及相关化学品残留量的测定 气相色谱–质谱法[S].
[4]GB/T 2405–2006 蒽醌[S].
[5]GB/T 19648–2006 蒽醌[S].
[6]叶碧莎,赵琳,宋悦华.保健食品中总蒽醌的测定[J].中国卫生检验杂志,2007,17 (5): 830–831.
[7]蔡晓,李宏.紫外分光光度法测定保健食品中总蒽醌的含量[J].中国食品卫生杂志,2007,19(1): 47–48.
[8]郭升平.高效液相色谱测定蒽醌[J].精细石油化工,1996(1): 59–60.
[9]文岛俊幸,本山总良,齐藤文孝,等.高效液相色谱法测定何首乌和夜交藤中蒽醌类成分的含量[J].药物分析杂志,1996,16 (4): 219–221.
[10]纪松岗,柴逸峰,吴玉田,等.超临界流体萃取一胶束电动毛细管色谱法分离测定大黄中蒽醌类组分的含量[J].分析化学研究简报,1998,26(11): 1 388–1 390.
[11]唐成国.气相色谱–质谱联用分析蒽醌工作液的组成[J].化学研究与利用,2000,12(3): 278–281.
[12]柴芸彬. GPC净化联合GC–MS–MS检测茶叶中的蒽醌[J].广州化工,2015,43 (4): 128–130.
[13]韩生银,代海香.降脂灵冲剂总黄硐及总蒽醌的测定[J].宁夏医学院学报,2001,23(2): 106–107.
[14]陈军,方芸,刁西辉.比色法测定逐瘀扶正胶囊中蒽醌类成分的含量[J].药学实践杂志,2002,20(5): 298 –300.
[15]沈伟健,余可垚,桂茜雯,等.分散固相萃取–气相色谱–串联质谱法测定蔬菜中107种农药的残留量[J].色谱,2009,27(4): 391–400.
[16]颜鸿飞,王美玲,陈红,等.固相萃取–气相色谱–串联质谱法测定茶叶中9种农药残留量[J].化学分析计量,2016,25(6): 62–66.
[17]惠恩健,盛振华,方文忠,等. QuEChERS 法联合在线GPC–GC–MS 测定独活中27种农药残留[J].中国实验方剂学杂志,2013,19(8): 138–141.
Determination of Anthraquinone in Tea by GC–MS–MS
He Zhenghe, Wei Bin, Wei Yunji, Zhu Zhenyi, He Jian, Feng Min, Zhang Ke, Qin Xian
[Huaian Entry–Exit Inspection and Quarantine Bureau, State Key Laboratory of Feed Safety Testing(Huaian), Huaian 223001, China]
The method for determination of anthraquinone in tea by gas chromatography–tandem mass spectrometry(GC–MS–MS) was established. The sample was extracted by acetonitrile of chromatographic purity, vortexed for 1 min, sonicated for 20 min, centrifuged and concentrated, then the extracts were purified by solid phase extraction(SPE). After being separated on DB–5MS capillary column, the target analyte was detected by GC–MS–MS under the multi reaction monitoring mode with a qualitative ion pair (m/z207.9, 152) and a quantitative ion pair (m/z207.9, 180), and quantified by the external standard method. The anthraquinone concentration and peak area of quantification ion had good relationship in the range of 1–500 μg/L, the correlation coefficient was 0.999 98. The detection limit was 1 μg/kg. The recoveries for 10, 20, 50 μg/kg spiked levels ranged from 92.85% to 98.10%, and the relative standard deviation of determination results was 8.50 %–11.05%(n=5). The method is suitable for determination of anthraquinone in tea with advantages of simple and rapid sample processing, low detection limit, high accuracy and precision.
tea; anthraquinone; solid phase extraction; GC–MS–MS
O657.7
A
1008–6145(2017)03–0018–04
*江苏出入境检验检疫局2016科研项目(2016KJ52)
联系人:何正和;E-mail: hezhenghehe@163.com
2017–02–12
10.3969/j.issn.1008–6145.2017.03.004
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
理化检验-化学分册(2020年12期)2021-01-26
新世纪智能(英语备考)(2018年11期)2018-12-29
中成药(2018年10期)2018-10-26
天然产物研究与开发(2018年8期)2018-09-10
食品界(2018年8期)2018-09-03
中成药(2018年4期)2018-04-26
中国环境监察(2016年7期)2016-10-23
中国现当代社会文化访谈录(2016年0期)2016-09-26
探测与控制学报(2015年4期)2015-12-15
