
犬瘟热病毒AH株全基因组序列测定及其分子特征
2017-06-05 15:19李丹丹宫晓华缪秋红李传峰陈宗艳张淼涛刘光清
中国动物传染病学报 2017年2期
刘 柱,李丹丹,李 琦,宫晓华,缪秋红,李传峰,陈宗艳,张淼涛,刘光清
(1.中国农业科学院上海兽医研究所,上海 200241;2.西北农林科技大学动物医学院,杨凌 712100;3. 甘肃农业大学动物医学院,兰州 730070;4.安徽农业大学动物科技学院,合肥 230036)
·研究论文·
犬瘟热病毒AH株全基因组序列测定及其分子特征
刘 柱1,2,李丹丹1,2,李 琦1,3,宫晓华1,4,缪秋红1,李传峰1,陈宗艳1,张淼涛2,刘光清1
(1.中国农业科学院上海兽医研究所,上海 200241;2.西北农林科技大学动物医学院,杨凌 712100;3. 甘肃农业大学动物医学院,兰州 730070;4.安徽农业大学动物科技学院,合肥 230036)
本研究对犬瘟热病毒(Canine distemper virus,CDV)安徽分离株(CDV-AH株)的全基因组序列进行了测定和分析。根据GenBank上已发表的部分CDV基因组全长序列设计了8对特异性引物,RT-PCR方法分段扩增出CDV-AH株的全基因组序列。采用DNAStar软件和MEGA软件对CDV-AH株与其他CDV毒株的基因序列进行了比对和分析,并绘制系统进化树。结果显示,CDV-AH株全长15 690 bp,编码6种结构蛋白(N、P、M、F、H和L),与CDV-PS株核苷酸序列同源性最高,为98.8%,而与CDV-1以及Onderstepoort等疫苗株的序列同源性较低,为92.1%。CDV-AH株在保守区域出现15处氨基酸突变,这些突变对其所编码蛋白结构和功能的影响需要进一步研究。基于H基因序列的系统进化分析表明,CDV-AH株与其他CDV野毒株共同形成一个拓扑群,属于亚洲I型。
犬瘟热病毒;全基因组;序列分析
犬瘟热(canine distemper,CD)是由犬瘟热病毒(Canine distemper virus,CDV)感染犬科或其他肉食动物造成的传染性极强的急性、病毒性传染病。CDV宿主范围较广泛,包括犬科、猫科、鼬科和浣熊科等肉食动物[1,2]。由于CDV对光和紫外线敏感,在0℃以上感染力迅速丧失,因此春秋和冬季是CD的多发季节,夏天发病率较低。虽然CDV对环境的抵抗力弱,但是动物一旦感染CDV,发病率和致死率都很高。宠物犬感染后发病率可达100%,死亡率也高达80%,许多经济动物如水貂等也可感染发病并给养殖业带来严重的经济损失[3],目前CD已成为危害毛皮动物的主要三种传染病之一。CDV也可感染一些珍稀动物,2014年12月,陕西省珍稀野生动物抢救饲养研究中心报道了圈养大熊猫感染CDV的病例,经检测确认此毒株是CDV强毒株,这是首次发现CDV强毒株感染大熊猫,由此表明CDV很有可能已发生变异,这对珍稀动物的保护敲响了警钟。此外,CDV具有种间传播能力,其宿主范围在不断扩大,目前发现日本猕猴也存在感染病例[4]。已有研究人员从Paget’s病的患者体内检出CDV核酸,这使得CD有可能成为继狂犬病之后,犬传播给人的第二种病毒性疾病[5]。
由于CDV感染性极强,同一饲养环境中一旦有动物感染并发病,即可造成全群动物发病,因而尽早进行疫苗注射是预防CD的根本。目前国内外CDV弱毒疫苗作为免疫防治的重要手段被广泛应用,但是近年来时有免疫动物感染CDV的报道。一方面可能是免疫失败造成的,另一方面可能是CDV发生变异产生了强毒株,从而使现有疫苗没有保护力,导致免疫动物发病。本研究对所分离毒株的全基因组进行了序列测序与遗传进化分析,结果将有助于我们了解目前流行毒株的遗传背景,也为进一步CDV亚单位疫苗和活载体疫苗的研制打下基础。
1 材料与方法
1.1 病料来源 来自安徽省合肥市某宠物医院。
1.2 主要试剂和仪器 总R NA提取试剂盒购自GeneMark公司;用于PCR扩增的酶、载体以及胶回收试剂盒均购自宝生物(大连)工程有限公司;RNA 加A尾试剂盒以及RNA纯化试剂盒均购自Ambion公司;SMARTer RACE 5'/3'试剂盒购自Clontech公司;全自动样品快速研磨仪购自上海净信科技有限公司;梯度PCR仪购自BioRad公司;微量分光光度计购自杭州奥盛仪器有限公司。
1.3 引物设计 利用DNAMAN软件,将GenBank上已经发表的部分CDV毒株基因组全长序列进行比对,在其保守区设计扩增引物,并且按照RACE试剂盒说明书要求,设计RACE特异性引物(表1)。引物由上海华津生物科技有限公司合成。
1.4 病毒RNA制备
1.4.1 研磨病料 取1.0 g 犬肺脏、肝脏等组织加液氮进行研磨,然后加入适量PBS于样品快速研磨仪中进行再次研磨,然后置于-80℃反复冻融3次,为病毒RNA的提取做准备。
1.4.2 病毒R NA的提取 根据GeneMark公司总RNA提取试剂盒说明书提取总RNA,测定浓度后,一部分-80℃保存备用,另一部分进行反转录,用于RTPCR反应。
1.5 RT-PCR反应 在无核酶污染的小离心管中加入 2 μ g RNA、1 μ L随机引物,加DEPC水至12.5 μ L,然后70℃反应10 min,立即冰浴2 min。离心数秒,使样品置于管底,加入M-MLV 反应缓冲液 5 μ L、dNTPs 5 μ L、核酸酶抑制剂0.5 μ L、M-MLV反转录酶2 μ L,构建25 μ L反应体系,37℃孵育60 min,最后75℃ 10 min灭活酶,获得cDNA模板。以此cDNA为模板,使用表1中特异性引物分别扩增FL1到FL8片段。PCR反应条件:95℃预变性3 min,94℃变性30 s,51℃退火1 min,根据片段长度来确定72℃延伸时间。胶回收PCR产物,分别将其克隆至pMD19-T载体。每个片段挑选3~4个克隆送上海华津生物技术有限公司测序。用DNAStar软件SeqMan工具将所测序列拼接成全长cDNA序列。

表1 本研究所用到的引物序列Table 1 Primers used in this study
1.6 5'和3'端真实序列测定 用Clontech公司SMARTer -RACE试剂盒来进行测定。
1.6.1 5'R ACE 根据1.5中所获取的全长cDNA片段,用Primer Premier 5.0软件进行分析,按照试剂盒说明书规定的引物设计原则,设计5'端特异性引物,引物序列见表1。然后进行RNA电泳验证RNA的完整性,当完整性良好时,再用随机引物反转录获得带有SMARTer IIA Oligonucleotide接头的cDNA,以5'端接头序列通用引物UPM以及所选择的最佳特异性引物作为上下游引物,PCR扩增目的基因。PCR体系:5'RACE-READY cDNA 2.5 μ L、10×UPM 5 μ L、5'GSP 1μ L、PCR-Grade H2O 15.5 μ L、2×SeqAmp Buffer 25 μ L、SeqAmp DNA Polymerase 1.0 μ L。反应条件:95℃预变性3 min;94℃变性30 s,68℃退火30 s,72℃延伸2 min,35个循环。
1.6.2 3'R ACE 引物的设计原则同5'RACE,首先根据要求设计3'端特异性引物,引物序列见表1。提取RNA,通过RNA电泳验证RNA完整性,由于CDV不含A尾,故首先进行加A尾操作。用Amibion公司加A尾试剂盒对病毒加A尾,再用RNA纯化试剂盒进行纯化。随后用3'RACE技术,用oligo 18反转录获得带有相应接头的cDNA,以3'端接头引物以及所选择的最佳特异性引物作为上下游引物,PCR扩增目的基因。PCR体系:3'RACE-READY cDNA 2.5 μ L、10×UPM 5μ L、3'GSP 1μ L、PCR-Grade H2O 15.5μ L、2×SeqAmp Buffer 25μ L、SeqAmp DNA Polymerase 1.0 μ L。反应条件:95℃预变性3 min,94℃变性30 s,68℃退火30 s,72℃延伸2 min,35个循环。
1.6.3 目的基因的克隆与鉴定 取以上PCR产物进行琼脂糖凝胶电泳鉴定,将与目的基因大小一致的片段进行胶回收,克隆至pMD-19T载体中,16℃连接过夜,再转化到trans-5α大肠杆菌感受态细胞中。12 h后挑菌,置于含有Amp抗性的LB培养基中过夜培养,菌液经PCR鉴定,挑选阳性菌液提取质粒,送至上海华津生物科技有限公司进行测序。
1.7 序列分析 使用DNAStar软件的SeqMan工具对所得序列进行拼接,找出序列开放阅读框数目及位置,利用MegAlign对获得的CDV-AH株基因组序列与GenBank上部分CDV基因全序列进行比较分析,找出该毒株基因组序列变异情况。用MEGA6.0软件来绘制进化树。
2 结果
2.1 RT-PCR结果以及RACE结果 以8对所设计的特异性引物用高保真DNA聚合酶扩增所需基因,获得8个目的片段(图1),通过RACE方法扩增5'端以及3'端目的基因片段(图2)。
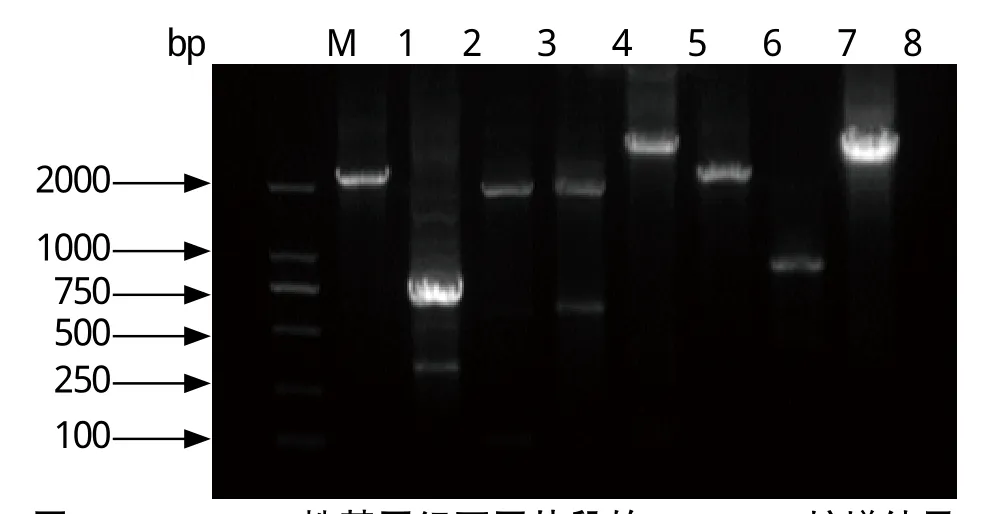
图1 CDV-AH株基因组不同片段的RT-PCR扩增结果Fig.1 Amplification results of the complete genome of CDV-AH strain by RT-PCRM: DNA分子量标准; 1~8: CDV-AH株1~8段的RT-PCR产物M: DNA Marker; 1-8: Genome fragments of CDV-AH strain
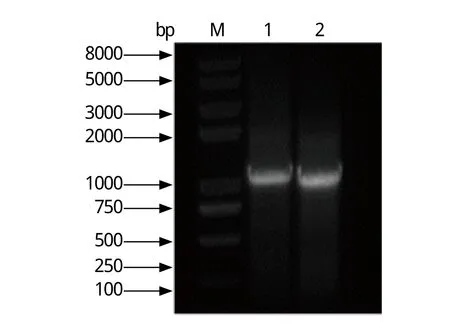
图2 3'RACE 和5'RACE扩增产物Fig.2 . Amplification products of 3'RACE and 5'RACEM: DNA分子量标准; 1: 3'RACE扩增产物; 2: 5'RACE扩增产物M: DNA Marker; 1: Amplification produt of 3'RACE; 2: Amplification product of 5'RACE
2.2 CDV-AH株基因组长度与基因排布 用DNAStar中的SeqMan工具将分段获取的CDV-AH基因组内部序列及基因组末端序列进行拼接,从而获得了CDVAH株全长基因组序列。将其登录至GenBank,登录号为KX 347928,大小为15 690 bp,与GenBank上公布的CDV全基因组大小一致。CDV-AH株基因组符合副粘病毒的基因组长度为6的倍数这一规律[6,7],该毒株的转录、复制与包装应该与其他副粘病毒一致。另外,通过DNAStar软件分析,CDVAH的整体结构也与其他已发表全序列CDV基因组结构类似,每个基因由3'端非编码区和5'端非编码区以及中间的编码区组成。由3'端到5'端分别为3'Leader序列、N基因、P基因、M基因、F基因、H基因、L基因、5'Trailer 序列,其大小分别为 52、1686、1658、1450、2209、1949、6645、41 nt(表2)。非编码区序列位于两个相邻的结构蛋白之间,含有一个半保守的多聚腺苷酸化和终止序列。N基因和P基因之间被121 nt非编码区间隔,P基因和M基因间被107 nt非编码区间隔,M基因和F基因被495 nt非编码区间隔,F基因和H基因间被155 nt非编码区间隔,H基因和L基因间被127 nt间隔。其中M基因和F基因之间的间隔序列最多,有研究表明M和F之间的基因间隔在调节F蛋白表达量和控制病毒的毒力方面起到重要作用[8]。另外基因间隔序列除了L基因 5'端尾随序列为CUA,其他非转录三联核苷酸标志均为CUU,猜测其基因间隔序列与转录信号有关,需要进一步研究验证。
2.3 CDV-AH株与参考毒株同源性比较 使用DNAStar软件包中的MegAlign工具对CDV-AH株全基因组序列进行比对分析,分析显示CDV-AH株与其他13株CDV毒株的核苷酸序列相似性为92.1%~98.8%。其中与CDV-PS株相似性最高,同源性为98.8%,而与CDV-1以及Onderstepoort疫苗株相似性相对较低,同源性都为92.1%,比对结果见表3。
2.4 CDV-AH株氨基酸序列分析
2.4.1 保守区域氨基酸序列分析 使用MegAlign软件比对CDV-AH株基因编码区和以上21株CDV毒株基因编码区氨基酸序列的异同,发现一些保守氨基酸发生了突变。CDV-AH株在编码区保守氨基酸处共发生15处突变,这些突变有可能会对蛋白的结构或功能产生或多或少的影响(表4)。

表2 CDV-AH株的基因组结构Table 2 Genome structure of CDV-AH strain

表3 CDV-AH与参考毒株全基因组序列核苷酸同源性分析Table 3 Analysis result of the nucleotide homology between CDV-AH strain and the reference CDV strains

表4 CDV-AH株在编码区保守氨基酸处氨基酸突变位点Table 4 The mutated sites in the coding region of CDVAH strain
2.4.2 CDV毒株H蛋白氨基酸序列分析 H蛋白作为病毒表面膜蛋白,在病毒感染过程中担负着重要的作用[9]。一方面,H蛋白识别并吸附于细胞表面受体,决定病毒细胞嗜性,进而决定病毒的宿主特异性;另一方面,蛋白协助病毒另一囊膜糖蛋白F蛋白,介导病毒囊膜与细胞膜发生膜融合,使病毒进入细胞。H蛋白极易发生变异,是CDV基因分型的重要依据[10],因此研究H蛋白的变异情况很有意义。图3中前5行为强毒株,后4行为疫苗株。通过对CDV各毒株H蛋白氨基酸序列分析,可以看出强毒株和疫苗株有明显的不同,疫苗株在某些固定的位点有固定的氨基酸序列,而强毒株在对应的位置上并没有规律性的变化,这些规律性的变化有可能是病毒在适应细胞过程中产生的变异,也有可能是病毒免疫原性位点。
H基因ORF为1824 nt,编码607 aa,通过基于H基因编码区氨基酸序列可以对CDV进行分型,在NCBI上查找一些典型CDV毒株H基因序列,运用MEGA 6.0软件对各毒株H基因编码区做聚类分析,得到CDV进化树,如图4所示,CDV-AH株属于Asia-I型,与疫苗株可信值较远,属于野毒株。
3 讨论
近年来CD频发,免疫失败比例逐渐增高。CDV属于RNA病毒,病毒RNA在复制过程中,其错误修复机制的酶活性很低,几乎没有,所以变异很快。因此对当前流行毒株全基因组测序分析很有必要。
本研究通过RT-PCR方法获得基因组8个末端重叠片段,又因为CDV基因组3'端和5'端对基因组转录和复制有重要作用,故使用RACE方法获得了准确的末端序列,得到准确的CDV全序列。序列分析显示,CDV-AH株基因组全长为15 690 nt,含有6个结构基因,N、P、M、F、H、L基因大小分别为1686、1658、1450、2209、1949、6645 nt。基因核苷酸变异遍布整个基因组,无论是基因间隔区还是基因编码区都存在着或多或少的变异,但是通过与其他CDV毒株的对比分析,各毒株基因组6个结构基因的起始和终止位置相似,长度也基本相同,并且其基因间隔序列也几乎一样,说明CDV基因组结构在遗传进化上存在一定保守性。通过与NCBI上21株CDV全基因组同源性分析,得知CDV-AH株与CDVPS株相似性最高,同源性为98.8%,而与CDV-1以及Onderstepoort疫苗株相似性相对较低,同源性为92.1%,其中CDV-PS株为强毒株。通过进化树分析显示,CDV-AH株属于亚洲I型,与疫苗株分属不同型。对CDV各强毒株和疫苗株的H基因所编码蛋白序列分析,发现疫苗株有一些固定氨基酸序列,而强毒株在固定位置无规律性。CDV-AH株符合强毒株规律,但是通过对CDV-AH株编码区所编码氨基酸序列进行分析,其在保守氨基酸处出现了15处突变,这些差异是否会影响病毒毒力,对其所编码蛋白的结构和功能有的影响还需要进一步研究。CDV作为一种古老的病毒,演化过程伴随着各种基因突变,CDV的宿主范围随着其演化过程不断扩大,很有可能是病毒通过自身突变对不同宿主环境有了进一步适应所导致。已有研究证实CDV的毒力与N蛋白密切相关,在病毒的装配、转录和复制过程中起调控作用[11],其本身保守性就很高,但是CDV-AH 株N蛋白发生1个氨基酸突变(第448处),由脯氨酸变为丝氨酸,这有可能影响到其所形成的二级结构,以及某些功能区的功能,进而造成N-RNA结构的稳定性下降,影响病毒基本生命活动。

图3 CDV各毒株H蛋白氨基酸序列分析Fig.3 Amino acid sequence alignment of the H protein of CDV strains注: CDV-AH、DQ191767、EU325724和FJ535063.1为强毒株; AB212966.1、AF305419.1、DQ903854.1 和 EU143737.1为疫苗株Note: CDV-AH, DQ191767, EU325724 and FJ535063.1 are the virulent strains; AB212966.1, AF305419.1, DQ903854.1 and EU143737.1 are the vaccine strains

图4 基于不同CDV毒株的H基因构建的遗传系统进化树Fig.4 Phylogenetic tree of CDV based on the amino acid sequence of H gene
CDV-AH在H蛋白保守氨基酸处也出现了1个突变,H蛋白是CDV重要毒力因子,其基因常作为CDV的分型依据。H蛋白作为病毒表面膜蛋白在病毒感染过程中担负着重要作用,可通过影响细胞嗜性的方式改变病毒毒力,具有很高的研究价值。安徽省接种过疫苗的犬又感染CDV,有可能是病毒变异造成的。不仅H基因发生变异,可能是全基因组发生各种变异共同造成,具体还需进一步调查研究证实。
CDV宿主较多,并且宿主范围不断扩大,甚至灵长目动物也可感染CDV,表明CDV具有感染人的潜在可能性,因此,构建反向遗传学平台来深入研究病毒的分子特征、致病机理以及病毒和宿主的关系显得尤为重要。同时,CDV具有做活载体疫苗的潜质,并已有成功先例。本研究通过对所分离到的CDV-AH株进行遗传学分析,为CDV活载体疫苗的开发提供了依据。
[1] A ppel M J, Y ates R A, Foley G L, et al. Canine distemper epizootic in lions, tigers, and leopards in North America[J]. J Vet Diagn Invest, 1994, 6(3): 277-288.
[2] Deem S L, Spelman L H, Y ates R A, et al. Canine distemper in terrestrial carnivores: a review[J]. J Zoo Wildl Med, 2000, 31(4): 441-451.
[3] Monne I, Fusaro A, Valastro V, et al. A distinct CDV genotype causing a major epidemic in Alpine wildlife[J]. Vet Microbiol, 2011, 150(1-2): 63-69.
[4] Sakai K, Y oshikawa T, Seki F, et al. Canine distemper virus associated with a lethal outbreak in monkeys can readily adapt to use human receptors[J]. J Virol, 2013, 87(12): 7170-7175.
[5] Mee A P, Dixon J A, Hoyland J A, et al. Detection of canine distemper virus in 100% of Paget's disease samples by in situ-reverse transcriptase-polymerase chain reaction[J]. Bone, 1998, 23(2): 171-175.
[6] Cherpillod P, Tipold A, Griot-Wenk M, et al. DNA vaccine encoding nucleocapsid and surface proteins of wild type canine distemper virus protects its natural host against distemper[J]. Vaccine, 2000, 18(26): 2927-2936.
[7] Bailey D, Banyard A, Dash P, et al. Full genome sequence of peste des petits ruminants virus, a member of the Morbillivirus genus[J]. Virus Res, 2005, 110(1-2): 119-124.
[8] Anderson D E, von Messling V. Region between the canine distemper virus M and F genes modulates virulence by controlling fusion protein expression[J]. J Virol, 2008, 82(21): 10510-10518.
[9] Bi Z, X ia X, Wang Y, et al. Development and characterization of neutralizing monoclonal antibodies against canine distemper virus hemagglutinin protein[J]. Microbiol Immunol, 2015, 59(4): 202-208.
[10] K e G M, Ho C H, Chiang M J, et al. Phylodynamic analysis of the canine distemper virus hemagglutinin gene[J]. BMC Vet Res, 2015, 11(1): 64.
[11] Peeters B P, Gruijthuijsen Y K, de Leeuw O S, et al. Genome replication of Newcastle disease virus: involvement of the rule-of-six[J]. Arch V irol, 2000, 145(9): 1829-1845.
GENOME SEQUENCING AND ANALYSIS OF CANINE DISTEMPER VIRUS STRAIN AH
LIU Zhu1,2, LI Dan-dan1,2, LI Qi1,3, GONG Xiao-hua1,4, MIAO Qiu-hong1, LI Chuan-feng1, CHEN Zong-yan1, ZHANG Miao-tao2, LIU Guang-qing1
(1. Shanghai Veterinary Research Institute, CAAS, Shanghai 200241, China; 2. College of Veterinary Medicine, Northwest A&F University, Yangling 712100, China; 3. College of Veterinary Medicine, Gansu Agricultural University, Lanzhou 730070, China; 4. College of Animal Sciences and Technology, Anhui Agricultural University, Hefei 230036, China)
The complete genome of Canine distemper virus (CDV) strain AH isolated from Anhui province was subcoloned and sequenced in the present study. Total 8 pairs of specifi c primers were designed according to the CDV reference for amplifi cation of the complete genome of CDV-AH strain in RT-PCR. Subsequently, the DNAStar software and MEGA software were used to compare and analyze the gene sequences of CDV-AH strain with reference strains. The results showed that total length of CDV-AH was 15 690 bp encoding 6 structural proteins (N P, M, F, H and L). The CDV-AH strain had 98.8% nucleotide sequence homology with CDV-PS strain and 92.1% with CDV-1 and Onderstepoort vaccine strains. The analysis of amino acid sequences showed 15 amino acid mutations in the conserved region of CDV-AH and the effect of these mutations on the protein structure and function need to be further studied. In addition, phylogenetic analysis of the CDV-AH strain and reference strains was carried out based on the H gene sequence. The resultsshowed that the CDV-AH strain and other CDV wild strains together came from a topological group belonging to the Asian I type.
Canine distemper virus; complete genome; sequence analysis
S852.659.5
A
1674-6422(2017)02-0001-08
2016-08-19
国家自然科学基金项目(31270194 , 31502068);上海市科技兴农重点攻关项目(2016043);上海市科委创新项目(13391901602);公益性农业科研专项(201303046);中央级公益性科研院所基本科研业务费专项(2016JB01,31101848)
刘柱,男,硕士研究生,预防兽医学专业
张 涛,E-mail:zmt6371@yahoo.com.cn;刘光清,E-mail:liugq@shvri.ac.cn
猜你喜欢
动物医学进展(2022年9期)2022-11-26
军事文摘(2022年16期)2022-08-24
科学大观园(2022年2期)2022-01-23
文萃报·周二版(2021年47期)2021-12-14
食品安全导刊(2021年21期)2021-08-30
今日农业(2021年11期)2021-08-13
湖南饲料(2021年3期)2021-07-28
兽医导刊(2020年13期)2020-12-31
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
