
旁路信号通路激活介导的EML4-ALK融合基因阳性肺癌细胞株H3122对alectinib继发耐药的研究*
2017-05-18 12:49李娅妮梁柳丹陈燕琼宋向群周韶璋
中国病理生理杂志 2017年5期
李娅妮, 梁柳丹, 陈燕琼, 宋向群, 周韶璋△
(广西医科大学 1研究生院, 2附属肿瘤医院呼吸肿瘤内科,广西 南宁 530021)
·论 著·
旁路信号通路激活介导的EML4-ALK融合基因阳性肺癌细胞株H3122对alectinib继发耐药的研究*
李娅妮1, 梁柳丹1, 陈燕琼1, 宋向群2, 周韶璋2△
(广西医科大学1研究生院,2附属肿瘤医院呼吸肿瘤内科,广西 南宁 530021)
目的: 本研究旨在探讨肝细胞生长因子(HGF)、表皮生长因子(EGF)及转化生长因子α(TGF-α)是否以旁路激活的方式诱导EML4-ALK融合基因阳性肺癌细胞株H3122对alectinib的耐药,并进一步探讨旁路信号激活在alectinib耐药中的作用。方法: 用不同浓度的alectinib、克唑替尼(crizotinib)、17-DMAG或(和)HGF(50 μg/L)、EGF(100 μg/L)、TGF-α(100 μg/L)处理EML4-ALK阳性肺癌细胞株H3122,采用CCK-8法检测细胞活力,流式细胞术检测细胞凋亡,应用Western blot技术检测细胞中ALK、c-Met、EGFR及相应磷酸化蛋白的表达,观察其下游通路关键蛋白AKT、ERK、p-AKT和p-ERK水平。结果: Alectinib作用72 h后,H3122细胞株的活力随着alectinib药物浓度的增加而逐渐下降,呈剂量依赖性。HGF、EGF和TGF-α诱导后,alectinib抑制H3122细胞的生长曲线往右移,HGF、EGF和TGF-α处理能够降低alectinib对肺癌细胞活力的抑制作用。0.05 μmol/L alectinib作用H3122细胞株48 h后的凋亡率为(20.12±1.36)%,而alectinib联合HGF、EGF和TGF-α后的凋亡率分别为(7.85±1.03)%、(5.60±0.79)%和(4.58±1.00)%,显著低于alectinib单药处理(P<0.05)。Alectinib单药成功抑制p-ALK及其下游信号通路,HGF明显增加细胞中p-Met及其下游p-AKT、p-ERK的蛋白水平,EGF和TGF-α明显增加细胞中p-EGFR及其下游p-AKT、p-ERK的表达,alectinib抑制p-ALK,但不能抑制HGF、EGF和TGF-α诱导的p-AKT和p-ERK的蛋白表达。此外,联合应用crizotinib和17-DMAG可以抑制因HGF和EGFR配体而导致的H3122耐药细胞的活力。结论: HGF、EGF和TGF-α可通过旁路激活的方式诱导EML4-ALK阳性肺癌细胞H3122对alectinib耐药,其机制可能与HGF激活c-Met磷酸化、EGF和TGF-α激活EGFR磷酸化有关。
EML4-ALK融合基因; Alectinib; 肝细胞生长因子; 表皮生长因子受体; 耐药性
棘皮动物微管相关蛋白样蛋白4-间变性淋巴瘤激酶(echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase,EML4-ALK)融合基因是非小细胞肺癌(non-small cell lung cancer,NSCLC)领域中一个重要的治疗靶点。ALK包含许多重要的生物学信号通路,影响肿瘤细胞的增殖、分化与凋亡[1]。 Alectinib是一种强效、选择性的二代ALK抑制剂,其对EML4-ALK阳性NSCLC有强的抑制作用,不仅抑制大多数与克唑替尼(crizotinib)耐药相关的ALK突变,并且对有中枢神经系统转移的患者有显著疗效[2],目前已被FDA批准用于克唑替尼耐药的ALK阳性NSCLC的治疗。然而最近在一些基础实验及ALK阳性患者中发现alectinib的耐药,目前已知的alectinib耐药机制中,ALK继发耐药突变[3]、小细胞肺癌的转变[4-5]以及上皮间质转化[6]等耐药方式都有了相应的解决方案,而旁路信号激活在alectinib耐药中的作用复杂多样,有待进一步研究。肝细胞生长因子(hepatocyte growth factor,HGF)是原癌基因c-Met的唯一配体,表皮生长因子(epidermal growth factor,EGF)及转化生长因子α(transforming growth factor-α,TGF-α)是表皮生长因子受体(epidermal growth factor receptor, EGFR)的配体,它们在与其相应的受体特异性结合后可激活一系列的跨膜信号通路(包括PI3K/AKT、MEK/ERK及STAT3),从而促进肿瘤的生长、侵袭和转移,常与临床预后差及耐药有关[7-8]。已有研究证实HGF通过激活c-Met信号通路诱导EGFR突变患者对EGFR-TKI耐药[9],不同于克唑替尼(为ALK及c-Met的双靶点抑制剂),alectinib对c-Met并没有抑制作用,c-Met信号通路可能是alectinib的耐药机制之一。另有研究报道EGFR信号旁路与克唑替尼耐药相关[10],但是它在alectinib耐药中的作用目前研究有待明确。本研究拟通过外源性加入HGF、EGF及TGF-α,比较和分析其对alectinib作用下肺癌细胞的生长和凋亡的影响,观察ALK、c-Met和EGFR信号通路的变化,旨在研究HGF、EGF及TGF-α旁路激活方式在诱导EMA4-ALK阳性NSCLC对alectinib耐药中的作用,并探索alectinib联合应用crizotinib和17-DMAG对H3122细胞活力的影响。
材 料 和 方 法
1 材料
1.1 细胞EML4-ALK融合基因阳性肺癌细胞株H3122购自科佰生物科技有限公司,含EML4-ALK融合基因变体1。
1.2 主要试剂与仪器 Alectinib和17-DMAG购于Selleckchem;crizotinib粉末制剂购于Cell Signaling Technology;胎牛血清和RPMI-1640培养基均购自BI;胰酶替代物购自Gibco;CCK-8细胞活力检测试剂盒购自Dojindo;Annexin V-PE/7-AAD细胞凋亡检测试剂盒购自BD;HGF、EGF和TGF-α购自PeproTech;抗GAPDH、ALK、p-ALK、c-Met、p-Met (Tyr1234/1235)、EGFR、p-EGFR(Tyr1068)、AKT、p-AKT(Ser473)、ERK、p-ERK(Thr202/Tyr204)单克隆抗体以及兔源和鼠源 II 抗均购自Cell Signaling Technology;Western blot实验仪器设备购自Bio-Rad。
2 方法
2.1 细胞培养及药物配制 将H3122细胞使用含10%灭活新生牛血清的RPMI-1640培养液,5% CO2、37 ℃的条件下培养,每3~4 d传代1次。Alectinib、crizotinib和17-DMAG原料用DMSO溶解制成106nmol/L的母液储存于-80 ℃冰箱,用药时用新鲜的RPMI-1640培养基稀释,并使DMSO的终浓度小于0.1%。HGF、EGF和TGF-α用无菌双离子水稀释成10 mg/L、20 mg/L和20 mg/L,分装储存于-20 ℃冰箱。
2.2 CCK-8实验检测细胞活力 取生长良好的对数期H3122细胞,每100 μL含细胞数为4×103个的细胞悬液接种于96孔板。待细胞贴壁后,吸去培养基,加入不同浓度的alectinib、crizotinib、17-DMAG或(和)50 μg/L HGF、100 μg/L EGF、100 μg/L TGF-α,以不加药孔为空白对照孔,以不含细胞,只含对应浓度药物培养基的孔为空白调零孔,设置4个复孔。作用72 h后,吸去培养基,加入100 μL含1/10体积的CCK-8溶液的培养基,继续孵育2~3 h后,用酶标仪测量波长450 nm处吸光度(A)值。细胞存活率(%)=(处理组平均A值-空白调零组平均A值)/(空白对照组平均A值-空白调零组平均A值)×100%。
2.3 流式细胞术凋亡检测 取生长良好的对数期H3122细胞,以每孔4×105个细胞数接种于6孔板中,待细胞贴壁后弃原培养基,实验分为4组:空白对照(control)组、alectinib处理组、重组细胞因子(HGF/EGF/TGF-α)处理组和alectinib联合重组细胞因子(HGF/EGF/TGF-α)处理组,按分组加入含0.05 μmol/L的alectinib或(和)50 μg/L HGF、100 μg/L EGF、100 μg/L TGF-α的培养基作用48 h。胰酶消化并分别收集各孔全部细胞,离心、弃上清液,冷生理盐水洗涤2次,按照细胞凋亡试剂盒说明书对细胞进行染色,避光、室温反应15 min后加入缓冲液,用流式细胞仪检测细胞凋亡。实验重复3次。
2.4 Western blot实验 实验分为4组:空白对照(control)组、alectinib处理组、重组细胞因子(HGF/EGF/TGF-α)处理组和alectinib联合重组细胞因子(HGF/EGF/TGF-α)处理组,先以0.05 μmol/L alectinib处理2 h,继而加入50 μg/L HGF、100 μg/L EGF或100 μg/L TGF-α刺激15 min,收集长满瓶的经不同处理的H3122细胞,按蛋白提取试剂说明书提取细胞总蛋白,所得总蛋白经10%聚丙烯酰胺凝胶电泳分离后,转移至PVDF膜,5%脱脂奶粉室温封闭1 h。分别使用稀释度为1∶1 000的ALK、1∶1 000的p-ALK、1∶1 000的c-Met、1∶1 000的p-Met(Tyr1234/1235)、1∶1 000的EGFR、1∶1 000的p-EGFR(Tyr1068)、1∶2 000的AKT 、1∶2 000的p-AKT(Ser473)、1∶1 000的ERK和1∶1 000的p-ERK(Thr202/Tyr204)抗体,4 ℃孵育过夜,TBST洗膜10 min,3次后用稀释度1∶2 000的兔鼠II抗室温下孵育1~2 h,ECL发光试剂盒显色,计算机扫描蛋白质条带,以条带灰度值确定蛋白表达,实验重复3次。
3 统计学处理
应用SPSS 17.0统计学软件,数值以均数±标准差(mean±SD)表示,两组间比较采用t检验, 多组数据比较采用单因素方差分析,以P<0.05表示差异有统计学意义。
结 果
1 CCK-8法检测alectinib单独或联合HGF/EGF/TGF-α处理对H3122细胞活力的影响
Alectinib作用72 h后,H3122细胞的活力随着alectinib药物浓度的升高相应下降,呈现显著的浓度依赖性抑制,alectinib作用H3122细胞72 h的IC50值为0.042 μmol/L。50 μg/L HGF处理后,alectinib作用于H3122细胞的IC50显著升高(P<0.05),而100 μg/L EGF或100 μg/L TGF-α处理后,H3122细胞对alectinib不敏感,给予100倍于IC50值的药物浓度仍未求出IC50,估计值远大于3 μmol/L。HGF(50 μg/L)、EGF(100 μg/L)和TGF-α(100 μg/L)诱导H3122细胞的药物浓度-细胞存活率曲线与非诱导曲线相比明显往右侧移。重组细胞生长因子单独处理H3122细胞出现不同程度促进细胞活力的作用,尤其是TGF-α对H3122细胞的促生长作用显著(P<0.05),见图1。
Figure 1.H3122 cell viability after treatment with various concentrations of alectinib alone or combined with HGF/EGF/TGF-α for 72 h detected by CCK-8 assay. Mean±SD.n=3.*P<0.05vsalectinib group.
图1 CCK-8法检测不同浓度alectinib单独或联合HGF/EGF/TGF-α作用72 h后H3122细胞的存活率
2 流式细胞术检测细胞凋亡
根据CCK-8法检测H3122细胞生长的情况,选择0.05 μmol/L alectinib单独或联合HGF/EGF/TGF-α处理H3122细胞48 h,检测细胞凋亡率。结果0.05 μmol/L alectinib单药处理48 h后H3122细胞的凋亡率为(20.12±1.36)%,50 μg/L HGF、100 μg/L EGF和100 μg/L TGF-α联合0.05 μmol/L alectinib处理48 h后的凋亡率分别为(7.85±1.03)%、(5.60±0.79)%和(4.58±1.00)%,显著低于alectinib单药处理(P<0.05)。实验结果表明alectinib对H3122细胞有促凋亡作用,HGF、EGF和TGF-α诱导作用可减少alectinib导致的H3122细胞凋亡,见图2。
3 Western blot检测alectinib单独或联合HGF/EGF/TGF-α处理对H3122细胞ALK、HGF/Met和EGFR信号通路蛋白表达的影响
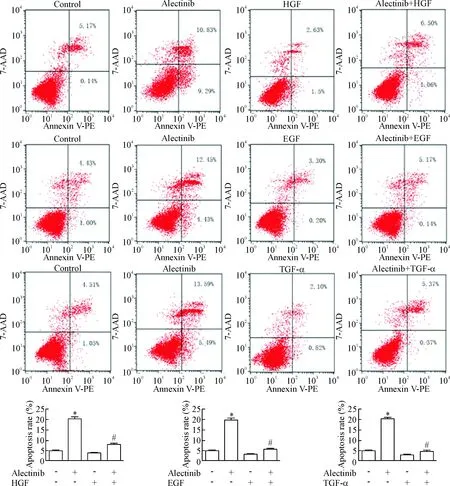
Figure 2.The apoptosis of H3122 cells treated with alectinib at 0.05 μmol/L alone or combined with HGF, EGF or TGF-α for 48 h. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsalectinib group.
图2 Alectinib单独或联合HGF、EGF和TGF-α作用 48 h对H3122细胞凋亡的情况
为了探索HGF、EGF和TGF-α旁路激活在诱导H3122细胞对alectinib耐药中的作用,我们采用Western blot技术检测ALK、c-Met、EGFR及相应磷酸化蛋白的表达,同时观察其下游通路关键蛋白AKT、ERK、p-AKT和p-ERK的水平。结果显示未经处理的H3122细胞可测得ALK和下游信号通路AKT、ERK及其磷酸化蛋白,H3122也表达c-Met及EGFR蛋白,但p-Met及p-EGFR蛋白测不出。0.05 μmol/L alectinib单药作用2 h后成功抑制ALK及其下游信号蛋白AKT、ERK的磷酸化,但对总蛋白的水平无明显影响。当联合重组细胞因子(50 μg/L HGF、100 μg/L EGF或100 μg/L TGF-α)同时作用于细胞时,alectinib虽然仍有效抑制ALK的磷酸化,但不能抑制下游信号蛋白AKT、ERK的磷酸化。HGF激活c-Met旁路途径、EGF和TGF-α激活EGFR旁路途径,可以不依赖ALK的活化而激活下游AKT、ERK的磷酸化。这些结果提示HGF通过激活c-Met旁路途径、EGF和TGF-α通过激活EGFR旁路途径进而激活下游通路来介导H3122细胞对alectinib的继发耐药,见图3。
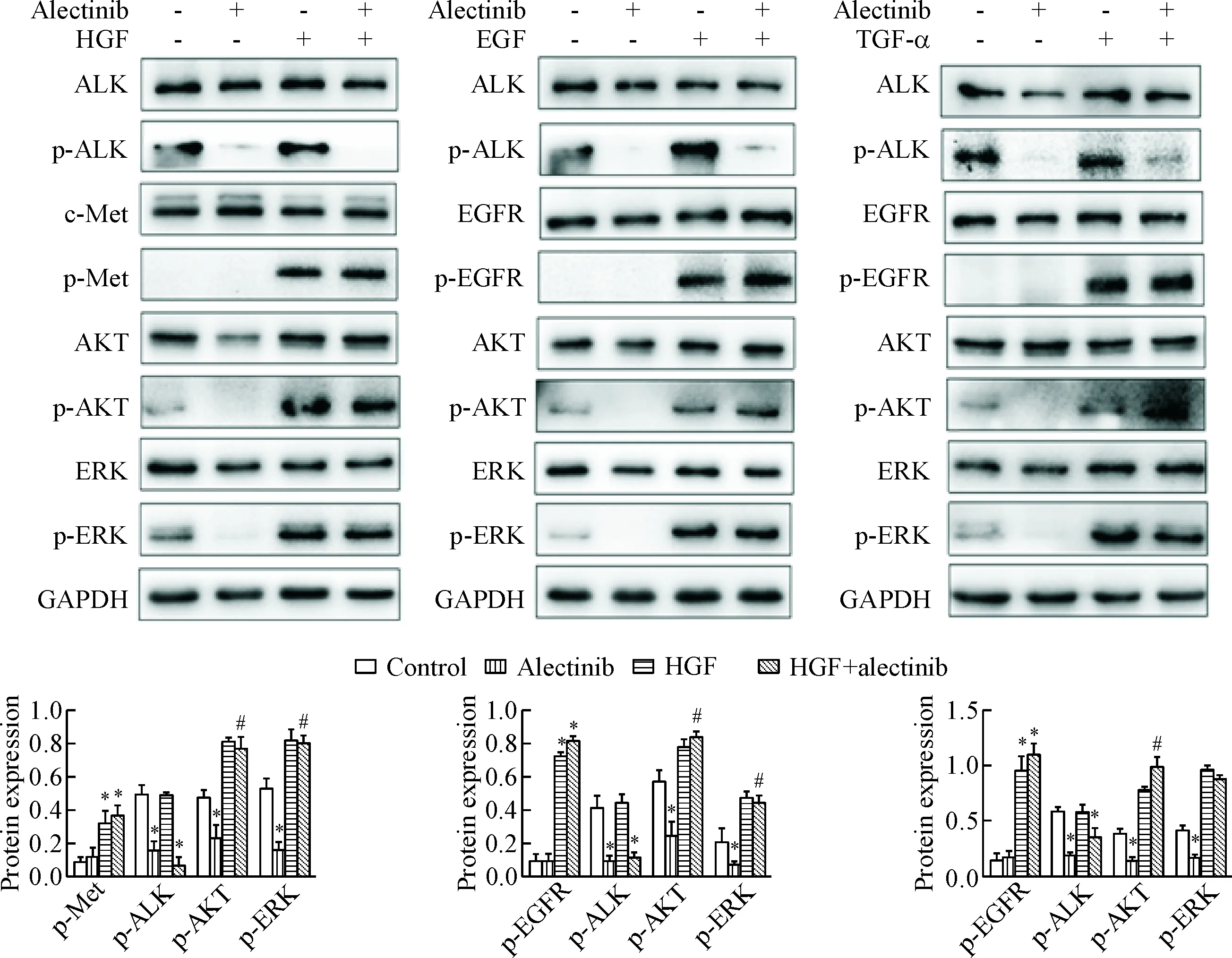
Figure 3.The protein levels in the H3122 cells treated with alectinib at 0.05 μmol/L alone or combined with HGF, EGF or TGF-α. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsalectinib group.
图3 Alectinib单独或联合HGF、EGF和TGF-α处理后H3122细胞的蛋白水平比较
4 Alectinib联合HGF/c-Met及热休克蛋白 90 (heat shock protein 90, Hsp90)抑制剂对HGF/EGF/TGF-α诱导的细胞增殖的影响
为进一步研究逆转由HGF/EGF/TGF-α介导的alectinib耐药,我们检测了alectinib联合crizotinib(HGF/c-Met抑制剂)和17-DMAG(Hsp90抑制剂)对H3122细胞活力的影响。结果发现0.1 μmol/L的crizotinib可解除HGF诱发的细胞对alectinib的耐药性变化,0.3 μmol/L 的17-DMAG 可以有效抑制H3122细胞在alectinib联合EGF/TGF-α处理下细胞的活力,见图4。
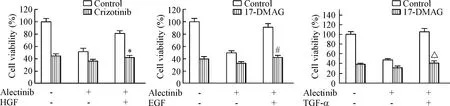
Figure 4.The effects of alectinib combined with crizotinib or 17-DMAG in the presence of HGF, EGF or TGF-α. Mean±SD.n=3.*P<0.05vsalectinib+HGF group;#P<0.05vsalectinib+EGF;△P<0.05vsalectinib+TGF-α group.
图4 Alectinib联合crizotinib或17-DMAG对HGF、EGF和TGF-α诱导的H3122细胞活力的影响
讨 论
Alectinib是第2代的ALK抑制剂,药物活性及选择性都高于第1代的克唑替尼,它对克唑替尼耐药的ALK阳性NSCLC及有中枢神经系统转移的患者均有效。但同其它靶向药物一样,在治疗一段时间后仍不可避免地发生耐药,探索其耐药的发生机制对于寻求克服方法具有重要意义。
ALK结合域二次突变[3]、细胞类型转变[4-5]以及上皮-间质转化[6]目前被认为是alectinib获得性耐药的重要机制。肿瘤细胞除了表达ALK外,同时还表达其它酪氨酸激酶活性的跨膜受体,称之为ALK旁路酪氨酸激酶信号。在肺癌患者中,MET扩增[11]、表皮生长因子受体2(human epidermal growth factor receptor-2,HER2)扩增[12]、HGF/c-Met[9]、生长停滞特异性基因产物6(growth arrest-specific gene 6,GAS6) /AXL[13]与胰岛素样生长因子1受体(human insulin-like growth factor 1 receptor,IGF-1R)[14]信号通路的激活,均可导致EGFR突变非小细胞肺癌患者对EGFR-TKI 的耐药。此外还发现HGF/c-Met[15]及IGF-1R[16]信号通路的激活参与了BRAF突变黑色素瘤患者对BRAF的耐药。有研究证实EGFR[10]、KIT/SCF[17]旁路信号通路的激活与克唑替尼耐药相关,但旁路信号激活在alectinib耐药发生发展中的作用目前尚未完全明确。
在我们的实验中,CCK-8细胞活力检测显示alectinib抑制EML4-ALK融合基因阳性肺癌细胞株H3122细胞的活力呈浓度依赖性,HGF、EGF及TGF-α诱导后IC50值显著升高。HGF、EGF及TGF-α诱导后药物浓度-细胞存活率曲线与非诱导曲线相比明显往右侧移。Alectinib促进细胞凋亡,HGF、EGF及TGF-α诱导后明显降低其促进细胞凋亡的作用。从IC50的变化、药物浓度-存活率曲线、细胞凋亡率的结果看出,HGF、EGF及TGF-α诱导H3122细胞对alectinib耐药。本研究检测出HGF诱导后增加细胞中p-Met及其下游信号通道蛋白,EGF、TGF-α诱导增加细胞中p-EGFR及其下游信号蛋白的水平,alectinib抑制p-ALK,但不能抑制由HGF、EGF和TGF-α诱导后p-AKT和p-ERK的蛋白水平。推测HGF激活c-Met磷酸化及EGF、TGF-α激活EGFR磷酸化,以旁路信号通路激活的方式诱导EML4-ALK阳性肺癌细胞株H3122对alectinib的耐药。既往有研究在药物浓度梯度递增法诱导的alectinib耐药株中检测出HGF、NRG1、EGF、IGF及TGF-α的mRNA高表达[18-20],提示HGF、NRG1,EGF、IGF及TGF-α可能是alectinib耐药的驱动因子,而本研究通过外源性加入生长因子的方法亦成功诱导H3122细胞对alectinib耐药,进一步证实了旁路激活在耐药中的作用。此外,NRG1/HER3的激活及IGF-1R也与alectinib耐药相关[18, 20],可见旁路信号激活诱导alectinib耐药的机制是复杂多样的,还需不断探索发现新的机制。
HGF、EGF及TGF-α虽然是本研究中介入的外源性生长因子,但在肿瘤细胞生长的微环境中也存在HGF、EGFR及IGF-1R等的配体,如内皮细胞产生的EGFR配体激活EGFR诱导克唑替尼及TAE684耐药,纤维细胞产生的HGF激活MET信号通路诱导TAE684的耐药[10]。有研究[20]报道了1例经alectinib治疗后继发耐药的患者,采用IHC检测患者用药前及耐药后的肿瘤活检标本,结果提示耐药后的标本中HGF呈现高表达,这些结果为我们的实验结果提供了临床依据。
有意思的是,我们的研究发现HGF/c-Met和EGFR信号通路的活化能够介导H3122细胞对alectinib的耐药,而克唑替尼作为ALK和c-Met的双靶点抑制剂可抑制因HGF诱发的耐药细胞的生长。EGFR是热休克蛋白90(HSP90)顾客蛋白之一,Hsp90是一种分子伴侣,它在调节许多顾客蛋白构象与功能的维持和调控方面发挥重要作用[21]。17-DMAG作为HSP90的抑制剂,对多种 NSCLC 细胞株具有抗增殖效应[22]。本研究中观察到alectinib联合应用17-DMAG可抑制由EGF、TGF-α诱导的H3122耐药细胞的增殖。这揭示了在ALK-TKI耐药患者中联合使用一代的ALK-TKI或其它TKI可能是潜在的克服耐药的方法,另有相关的研究也报道了L1198F突变导致的三代ALK-TKI Lorlatinib耐药患者再次用回克唑替尼并从中获益[23],这似乎在演绎着ALK-TKI的轮回,如果在多线进展的肿瘤细胞中发现新的耐药机制,联合使用相关信号通路抑制剂可能仍敏感有效。我们研究alectinib的耐药机制,为联合应用靶向受体配体抑制剂克服alectinib耐药提供实验室依据,为临床耐药患者提供了新的治疗策略。
本研究证实外源性加入HGF、EGF和TGF-α可以诱导EML4-ALK阳性细胞H3122对alectinib的耐药,并揭示了HGF/c-Met和EGFR旁路信号通路的活化可能是其耐药机制,这为今后深入研究针对EML4-ALK靶点的靶向耐药和逆转耐药机制提供分子生物学基础。然而,该研究仅限在细胞水平,仍需更多的临床耐药患者组织标本证实,可望更好阐明 HGF/c-Met和EGFR旁路信号通路激活在alectinib耐药中的作用。
[1] 戴 辉, 宋向群, 潘星辰, 等. mTOR 信号通路在克唑替尼诱导的EML4-ALK融合基因阳性肺癌细胞株 H2228凋亡中的作用[J]. 中国病理生理杂志, 2014, 30(6):1103-1109.
[2] Gadgeel SM, Gandhi L, Riely GJ, et al. Safety and acti-vity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study[J]. Lancet Oncol, 2014, 15(10):1119-1128.
[3] Katayama R, Friboulet L, Koike S, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib[J]. Clin Cancer Res, 2014, 20(22):5686-5696.
[4] Takegawa N, Hayashi H, Iizuka N, et al. Transformation of ALK rearrangement-positive adenocarcinoma to small-cell lung cancer in association with acquired resistance to alectinib[J]. Ann Oncol, 2016, 27(5):953-955.
[5] Fujita S, Masago K, Katakami N, et al. Transformation to SCLC after treatment with the ALK inhibitor alectinib[J]. J Thorac Oncol, 2016, 11(6):e67-e72.
[6] Kogita A, Togashi Y, Hayashi H, et al. Hypoxia induces resistance to ALK inhibitors in the H3122 non-small cell lung cancer cell line with an ALK rearrangement via epithelial-mesenchymal transition[J]. Int J Oncol, 2014, 45(4):1430-1436.
[7] Corso S, Giordano S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: from basic research to a clinical perspective[J]. Cancer Discov, 2013, 3(9):978-992.
[8] Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer[J]. J Clin Oncol, 2013, 31(8):1089-1096.
[9] Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations[J]. Cancer Res, 2008, 68(22):9479-9487.
[10]Yamada T, Takeuchi S, Nakade J, et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells[J]. Clin Cancer Res, 2012, 18(13):3592-3602.
[11]Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling[J]. Science, 2007, 316(5827):1039-1043.
[12]Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation[J]. Cancer Discov, 2012, 2(10):922-933.
[13]Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer[J]. Nat Genet, 2012, 44(8):8528-8560.
[14]Yeo CD, Park KH, Park CK, et al. Expression of insulin-like growth factor 1 receptor (IGF-1R) predicts poor responses to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer patients harboring activating EGFR mutations[J]. Lung Cancer, 2015, 87(3):311-317.
[15]Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion[J]. Nature, 2012, 487(7408):500-504.
[16]Hilmi C, Larribere L, Giuliano S, et al. IGF1 promotes resistance to apoptosis in melanoma cells through an increased expression of BCL2, BCL-X(L), and survivin[J]. J Invest Dermatol, 2008, 128(6):1499-1505.
[17]Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers[J]. Sci Transl Med, 2012, 4(120):120ra17.
[18]Dong X, Fernandez-Salas E, Li E, et al. Elucidation of resistance mechanisms to second-generation ALK inhibitors alectinib and ceritinib in non-small cell lung cancer cells[J]. Neoplasia, 2016, 18(3):162-171.
[19]Tani T, Yasuda H, Hamamoto J, et al. Activation of EGFR bypass signaling by TGFα overexpression induces acquired resistance to alectinib in ALK-translocated lung cancer cells[J]. Mol Cancer Ther, 2016,15(1):162-171.
[20]Isozaki H, Ichihara E, Takigawa N, et al. Non-small cell lung cancer cells acquire resistance to the ALK inhibitor alectinib by activating alternative receptor tyrosine kinases[J]. Cancer Res, 2016,76(6):1506-1516.
[21]Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights[J]. Nat Rev Mol Cell Biol, 2010, 11(7):515-528.
[22]Kobayashi N, Toyooka S, Soh J, et al. The anti-proliferative effect of heat shock protein 90 inhibitor, 17-DMAG, on non-small-cell lung cancers being resistant to EGFR tyrosine kinase inhibitor[J]. Lung Cancer, 2012, 75(2):161-166.
[23]Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F[J]. N Engl J Med, 2016, 374(1):54-61.
(责任编辑: 陈妙玲, 罗 森)
Bypass signaling pathway activation mediates resistance ofEML4-ALKfusion gene positive lung cancer cell line H3122 to alectinib
LI Ya-ni1, LIANG Liu-dan1, CHEN Yan-qiong1, SONG Xiang-qun2, ZHOU Shao-zhang2
(1PostgraduateCollege,2DepartmentofRespiratoryOncology,AffiliatedTumorHospital,GuangxiMedicalUniversity,Nanning530021,China.E-mail:zhoushaozhang@qq.com)
AIM: To detect the changes of active status of bypass signaling pathways inEML4-ALKpositive lung cancer cell line H3122 treated with alectinib, hepatocyte growth factor (HGF), epidermal growth factor (EGF) and transforming growth factor-α (TGF-α), and to explore the potential mechanisms. METHODS:EML4-ALKpositive cell line H3122 was treated with increasing concentrations of alectinib or/and induced by HGF, EGF and TGF-α. The cell viability was measured by CCK-8 assay. The cell apoptosis was analyzed by flow cytometry. The protein levels and phosphorylation status of ALK, c-Met and EGFR, and the downstream molecules AKT, ERK, p-AKT and p-ERK were examined by Western blot. RESULTS: The viability of the H3122 cells was inhibited by alectinib in a dose-dependent manner after administrated for 72 h, and the IC50value was 0.042 μmol/L. The concentration-growth curves of the H3122 cells shifted to the right after induced by HGF, EGF and TGF-α. After treatment with alectinib at 0.05 μmol/L for 48 h, the apoptotic rate of H3122 cells was (20.12±1.36)%, while the apoptotic rates of the cells in the groups of alectinib combined with HGF, EGF or TGF-α were (7.85±1.03)%, (5.60±0.79)% and (4.58±1.00)%, respectively. Those values were remarkably lower than those in alectinib single treatment group (P<0.05). Alectinib inhibited the protein levels of p-ALK and its downstream signaling pathway molecules, while HGF significantly up-regulated the protein levels of p-Met and its downstream p-AKT and p-ERK. Besides, EGF and TGF-α remarkablely up-regulated the protein levels of p-EGFR and its downstream p-AKT and p-ERK. Combined treatment with crizotinib and 17-DMAG successfully inhibited the viability of the H3122 cells even in the presence of the HGF and EGFR ligands, respectively. CONCLUSION: Bypass signaling pathways are activated by HGF, EGF and TGF-α inEML4-ALKpositive lung cancer cell line H3122, which may be linked to alectinib resistance.
EML4-ALKfusion gene; Alectinib; Hepatocyte growth factor; Epidermal growth factor receptor; Drug resistance
1000- 4718(2017)05- 0769- 07
2016- 11- 28
2017- 03- 17
国家自然科学基金资助项目(No. 81260357; No. 81060188); 广西自然科学基金资助项目(No. 2015GXNSFAA139162)
R730.23
A
10.3969/j.issn.1000- 4718.2017.05.001
杂志网址: http://www.cjpp.net
△通讯作者 Tel: 0771-5334955; E-mail: zhoushaozhang@qq.com
猜你喜欢
保健医苑(2022年5期)2022-06-10
现代临床医学(2022年3期)2022-06-06
中国典型病例大全(2022年9期)2022-04-19
昆明医科大学学报(2022年1期)2022-02-28
保健与生活(2021年24期)2021-12-12
建材发展导向(2021年16期)2021-10-12
中国现代医药杂志(2020年10期)2020-12-14
科学大众(2020年12期)2020-08-13
发电设备(2020年1期)2020-02-12
医学新知(2019年4期)2020-01-02
