
单核细胞增生性李斯特菌inlA和inlB基因缺失菌株的构建
2017-04-27 07:52:30刘武康陈国薇丁承超谢曼曼
微生物学杂志 2017年1期
刘武康, 李 森, 陈国薇, 罗 勤, 吴 嫚, 丁承超, 谢曼曼, 刘 箐 *
(1.上海理工大学 医疗器械与食品学院,上海 200093;2.华中师范大学 生命科学学院,湖北 武汉 430079)
单核细胞增生性李斯特菌inlA和inlB基因缺失菌株的构建
刘武康1, 李 森1, 陈国薇1, 罗 勤2, 吴 嫚1, 丁承超1, 谢曼曼1, 刘 箐1*
(1.上海理工大学 医疗器械与食品学院,上海 200093;2.华中师范大学 生命科学学院,湖北 武汉 430079)
单核细胞增生性李斯特菌(Listeriamonocytogenes,LM)是人畜共患的食源性致病菌,其对恶劣环境有较强的抵抗力,广泛存在于各种食品加工环境,容易引起严重的食品安全问题。LM是一种胞内寄生菌,其毒力因子内化素A(internalin A,InlA)和内化素B(internalin B,InlB)被认为在LM穿透宿主屏障、入侵宿主细胞以及胞间传播等过程中起到重要作用。本文利用同源重组法将LM野生菌株EGDe的inlA和inlB基因的敲除,对inlA和inlB基因缺失菌株的基本生物学特性进行研究,并运用RealTime-PCR监测LM的毒力基因表达。实验结果表明,基因的缺失对突变菌株的生长以及对环境中的氯化钠(NaCl)和乙醇(EtOH)的耐受能力没有影响,但多个毒力基因的表达量明显下降。该缺失菌株的构建为进一步研究InlA和InlB在LM入侵宿主细胞过程中的具体功能提供了重要材料。
单核细胞增生性李斯特菌;内化素A;内化素B;基因敲除;RealTime-PCR
单核细胞增生性李斯特菌(Listeriamonocytogenes,LM)是一种革兰阳性、腐生的人畜共患食源性致病菌,在土壤和腐败植被中大量分布[1]。感染LM的患者致死率可达20%~30%,死亡病例中以儿童、老人、孕妇等居多,因此LM是毒力最强的食源性致病菌之一[2]。LM对外界生存环境的适应能力快,对恶劣的生存条件如极寒极热、高渗、强酸强碱等都有较强的抵抗能力,同时LM能生成生物被膜,既能帮助其抵抗外界环境中的生存压力,又使其能长期生存在食品加工、运输等过程中所使用的工具和器皿上,带来极大的食品质量安全风险[3]。LM感染性很强,可以穿透宿主的肠道、胎盘和血脑三大屏障,在宿主细胞内寄生、繁殖并在胞间传播,LM感染宿主的过程中每一个步骤都需要多个毒力基因共同协作完成,LM感染宿主的过程可大致概括为内化进入细胞、逃逸吞噬、胞质内移动并在胞间传播三个主要阶段[4]。内化素蛋白家族便是LM众多毒力因子中比较重要的一类,它们在介导LM内化进入宿主非吞噬细胞的过程中有着非常重要的作用[5]。InlA和InlB是内化素蛋白家族中最重要的内化素蛋白,是最早被发现的介导LM入侵宿主细胞的内化素,也是迄今为止研究最为深入的LM的毒力因子之一。这两种蛋白的结构特点:在N-末端具有相似的结构,如信号肽区域,富含亮氨酸重复区域 (Leucine-rich repeat domain,LRR)等,但InlA 的C-末端的分选信号(Sorting signal)区域决定其通过共价结合的方式固定在细菌的细胞壁上,而InlB的C-末端富含甘氨酸/色氨酸的GW基元使其通过静电作用与细胞壁中脂磷壁酸结合,同时由于InlB的N-末端具有分泌信号, 因此也可能被分泌到LM 的胞外[6]。为进一步探究InlA、InlB对LM毒力的影响,本研究使用温敏型穿梭质粒pKSV7运用同源重组技术构建了inlA和inlB基因缺失的LM基因缺失菌株[7-8]。inlA和inlB基因缺失LM菌株的构建为深入研究该毒力基因在LM侵袭宿主细胞过程中的具体功能提供了参考。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒 血清型为1/2 a单核细胞增生李斯特菌的野生型菌株EGDe、穿梭质粒pKSV7 (大肠埃希菌中为氨苄青霉素抗性,LM中为氯霉素抗性)质粒由华中师范大学罗勤博士馈赠;大肠埃希菌(Escherichiacoli)JM109感受态细胞和pMD19-T Vector 购于大连宝生物(TaKaRa)公司。
1.1.2 培养基和试剂 本研究使用的脑心浸液培养基(BHI)、胰蛋白胨、酵母浸粉购于北京陆桥技术股份有限公司;Taq酶、dNTPS、反转录试剂盒等购于大连宝生物(TaKaRa)公司;引物由生工生物工程(上海)股份有限公司合成;细菌基因组抽提试剂盒、核酸凝胶回收试剂盒、质粒抽提试剂盒购于天根生化科技(北京)有限公司;其他化学试剂均为分析纯。
1.1.3 主要仪器 9500型PCR仪和7500型实时荧光定量PCR仪购自美国应用生物公司(ABI);SpectraMax M2型多功能全波长酶标仪购自美国分子设备公司(Molecular Devices);核酸电泳设备、电转化仪、凝胶成像仪及相关软件购自美国伯乐公司(Bio-rad)。
1.2 方法
1.2.1 引物序列 基因敲除操作过程中所使用的引物根据美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)对LM相关基因序列的报导使用DNAman软件自行设计,引物序列的详细信息见表1。A1、A2用于扩增inlA的上游片段,A3、A4用于扩增inlA的下游片段,A5、A6用于扩增inlA编码区内的片段而鉴定基因是否缺失;B1、B2用于扩增inlB的上游片段,B3、B4用于扩增inlB的下游片段,B5、B6用于扩增inlB编码区内的片段而鉴定基因是否缺失。引物A1、B1中含有SmaI的酶切位点,A4、B4中含有PstI的酶切位点,A2、A3以及B2、B3中均含有XbaI的酶切位点。
RealTime-PCR所使用的相关引物由本实验室其他人员前期设计,经验证特异性良好,此处不再列出详细序列信息。

表1 引物序列
1.2.2 LM基因组DNA和总RNA的提取 LM基因组DNA的提取使用细菌基因组抽提试剂盒,详细操作步骤见试剂盒说明书。LM总RNA提取方法:取过夜培养的菌液离心收集菌体,用适量溶菌酶在37 ℃温浴30 min左右,使用Trizol破碎菌体后加入氯仿(三氯甲烷)震荡混匀,离心后转移上层水相,加入异丙醇使RNA沉淀,收集沉淀,用适量75%乙醇清洗后用适量无RNase的无菌水溶解沉淀。
1.2.3inlA和inlB基因上下游同源臂的扩增及连接 以LM基因组DNA为模板进行PCR扩增,PCR产物进行琼脂糖凝胶电泳,并割胶回收,割胶回收具体操作见核酸凝胶回收试剂盒说明书。上下游同源臂单酶切并进行电泳、割胶回收,用T4 DNA连接酶过夜连接,连接完成后进行T-A克隆并导入JM109感受态细胞中,筛选阳性克隆保存。
1.2.4 pKSV7重组质粒的构建 将连有同源臂的T载体与pKSV7质粒使用相应的限制性内切酶进行双酶切,电泳后割胶回收,使用T4 DNA连接酶过夜连接并导入JM109感受态细胞中。将阳性克隆的质粒抽提后进行双酶切鉴定并测序,测序由华大基因完成,测序后将序列比对正确的质粒保存待后续实验使用。
1.2.5 重组质粒电转化LM感受态细胞 根据Park等[9]的报导,用终浓度为5 μg/mL的青霉素G和3.5×SMHEM溶液(952 mmol/L 蔗糖、3.5 mmol/L MgCl2和7 mmol/L HEPES溶于50 mL双蒸水,使用0.22 μm滤膜过滤除菌)处理菌体,分装至小离心管中于-80 ℃保存。电转场强设置为11.25~12.5 kV,电击时间为3~4 ms。
1.2.6inlA和inlB基因缺失突变株的筛选与鉴定 将含有重组质粒的LM转接到BHI(含10 μg/mL Cam)液体培养基中41 ℃连续培养并转接8~10次,将末代细菌培养物划线于BHI(含10 μg/mL Cam)平板上 41 ℃过夜培养;挑取BHI(含10 μg/mL Cam)平板上的单菌落于BHI液体培养基中30 ℃连续培养并转接6~8次,取末代培养物划线于BHI平板,30 ℃过夜培养;挑取单菌落进行PCR鉴定,并将疑似突变株分别划线于抗性平板和非抗性平板;对突变区域的基因序列测序以验证基因完全缺失。
1.2.7 突变株生长曲线及NaCl、EtOH耐受能力的测定 将LM、LM△inlA和LM△inlB接种于BHI液体培养基中过夜培养,次日取1 mL饱和菌液转接到100 mL新鲜BHI液体培养基中,200 r/min振荡培养,每隔1 h取200 μL菌液加入96孔板中测定OD600,共测定12 h,实验设有3个平行组并重复3次。对菌株NaCl、EtOH耐受能力测定时,将培养基改为含有质量分数5% NaCl的BHI液体培养基和含有体积分数3% EtOH的BHI液体培养基,其他操作同生长曲线的测定。
1.2.8 RealTime-PCR 将提取的LM、LM△inlA和LM△inlB总RNA反转录获得cDNA(反转录试剂盒中含有去除基因组DNA的相关试剂),以cDNA为模板进行SYBR Green荧光染料法RealTime-PCR,以LM的16S 核糖体RNA为内参基因,测定LM的11个主要毒力基因表达变化。
1.2.9 数据处理 本研究所有电泳图片使用Adobe PhotoShop CS6处理,实验数据使用GraphPad Prism5处理并绘制图表。
2 结果与分析
2.1inlA和inlB基因上下游同源臂的扩增
同源臂扩增结果见图1,inlA基因上游同源臂790 bp(1、2泳道),下游同源臂873 bp(3、4泳道);inlB基因上游同源臂747 bp(1、2泳道),下游同源臂607 bp(3、4泳道),扩增片段与预期片段大小相符且特异性良好无杂带。同时确定4对引物进行PCR反应时的退火温度均为56 ℃。
2.2 pKSV7重组质粒的构建
含有重组质粒的阳性克隆筛选、鉴定结果见图2,A、B图中1号泳道均为以无菌水为扩增模板的阴性对照,2~5号泳道为鉴定样本。电泳图显示重组质粒构建成功并筛选到阳性克隆,双酶切鉴定与测序结果表明,重组质粒上连接的目的基因同源臂序列与NCBI 报导的LM相关基因序列匹配度达到99%以上,无大量突变和错配情况存在,可以用于后续实验。
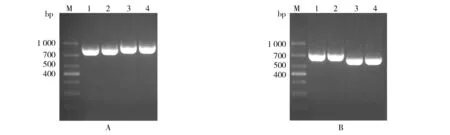
图1 上下游同源臂扩增Fig.1 Amplification of the flanking sequencesA:inlA基因上下游同源臂的扩增;B:inlB基因上下游同源臂的扩增;M:DL1 000 bp MarkerA: Amplification of the flanking sequences of inlA; B: Amplification of the flanking sequences of inlB; M: DL1 000 bp Marker

图2 重组质粒鉴定Fig.2 Identification of recombinant plasmidA:inlA基因重组质粒鉴定;B:inlB基因重组质粒鉴定;M:500 bp DNA Ladder MarkerA: Identification of recombinant plasmid of inlA;B: Identification of recombinant plasmid of inlB; M: 500 bp DNA Ladder Marker
2.3inlA和inlB基因缺失突变株的筛选与鉴定
图3A为inlA基因缺失菌株的筛选结果,1号泳道为以无菌水为扩增模板的阴性对照,2号泳道为以LM基因组DNA作为模板的阳性对照,电泳结果显示,4、5号泳道未扩增出目标片段(448 bp),说明该样本为疑似突变株。测序结果表明样本中的inlA基因已经缺失,而抗性鉴定显示,基因缺失菌株完全失去氯霉素抗性,将该菌株命名为LM△inlA,菌液冻存用于后续实验。
图3B为inlB基因缺失菌株的筛选结果,1号泳道为以LM基因组DNA作为模板的阳性对照,2号泳道为以无菌水为扩增模板的阴性对照,电泳结果显示,仅有6号泳道未扩增出目标片段(560 bp),说明该样本为疑似突变株。测序结果表明样本中的inlB基因已经缺失,抗性鉴定显示基因缺失菌株完全失去氯霉素抗性,将inlB基因缺失的LM菌株命名为LM△inlB,菌液冻存用于后续实验。
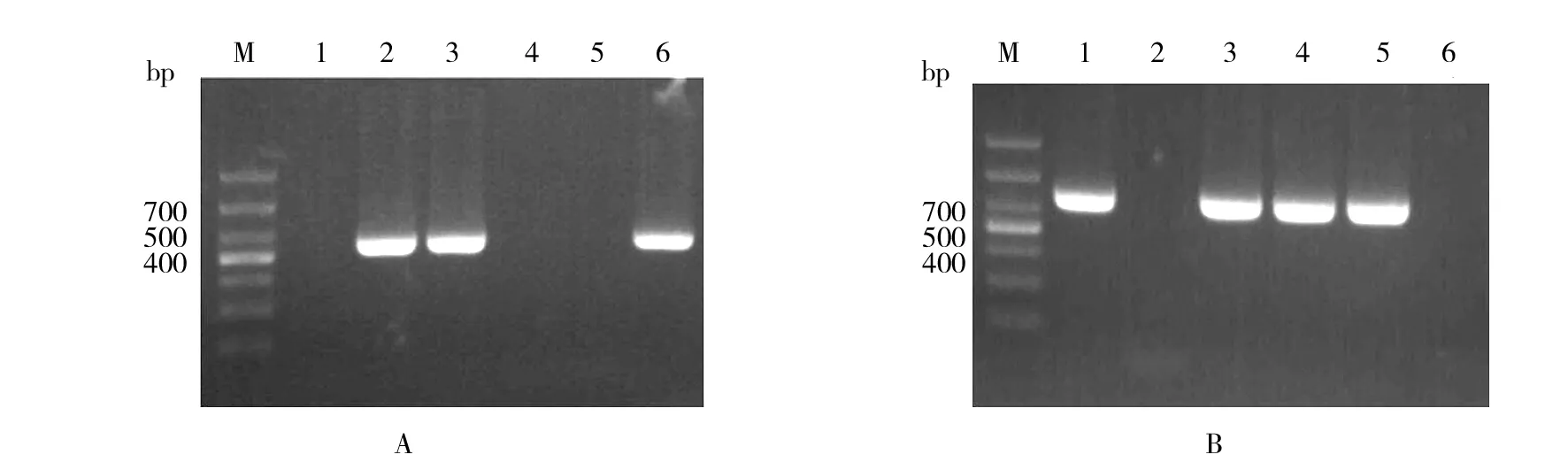
图3 基因缺失菌株的筛选Fig.3 Identification of the deletion mutantA:inlA基因缺失菌株鉴定;B:inlB基因缺失菌株鉴定;M:DL1 000 bp MarkerA: Identification of the mutant in inlA;B: Identification of the mutant in inlB;M:DL1 000 bp Marker
2.4 突变株生长曲线和NaCl、EtOH耐受能力的测定
LM、LM△inlA和LM△inlB三株菌株的生长曲线以及NaCl、EtOH耐受能力的测定完成后,以测定时间为横坐标,OD600为纵坐标绘制曲线。结果如图4所示,inlA和inlB基因的缺失对LM的生长和NaCl、EtOH的耐受能力没有显著影响,所以inlA和inlB 很可能不是LM的正常生长以及抵御不良生长环境的过程中必需的基因。由于LM△inlA和LM△inlB的生长能力没有改变,后续针对突变菌株毒力、致病性等方面的研究则无需考虑菌株自身生长能力的差异带来的影响。

图4 突变株生长曲线和NaCl、EtOH耐受能力测定Fig.4 Growth curvature and NaCl,ethanol tolerance of mutant strainA:生长曲线测定;B:5%NaCl耐受能力的测定;C:3%EtOH耐受能力的测定A:Growth curvature; B:5% NaCl tolerance;C:3% ethanol tolerance
2.5 RealTime-PCR
LM△inlA和LM△inlB的毒力基因RealTime-PCR结果如图5所示,LM△inlA和LM△inlB中的hly、plcA、prfA、sigB和srtA均有明显下调(P<0.05),2-△△Ct均在0.5左右,即基因的表达下调一半左右;LM△inlB中还有inlA、inlC、plcB、vip基因有明显下调(P<0.05);LM△inlA和LM△inlB中actA基因的表达量都没有明显变化。

图5 LM毒力基因RealTime-PCRFig.5 RealTime-PCR of virulence genes of LM
3 讨 论
本研究运用同源重组技术,成功地对单核细胞增生性李斯特菌野生型菌株EGDe进行了inlA和inlB基因的敲除,同时对突变株的生长、抗胁迫能力进行了研究,还利用RealTime-PCR技术测定了突变株中LM的主要毒力基因表达变化,为深入研究InlA和InlB在介导LM入侵宿主细胞过程中的具体作用机制提供了参考。
本研究使用的基因改造方法为使用温敏型自杀载体pKSV7,利用载体在41 ℃和氯霉素双重压力下整合进入LM基因组DNA进行复制的特点,将目的基因上下游同源臂连接于载体上,引导载体在菌体内部特定位置进行同源重组,从而实现目的基因的敲除。此种方法有操作简单、只需基础的分子生物学实验设备即可进行的优点,但与目前新兴的多种基因编辑技术相比,存在实验周期长、依赖专门的穿梭载体、筛选过程中获得突变株的概率不高等明显的缺点。目前,将最新的基因编辑技术应用于LM的基因改造还鲜有报导,该领域若能取得突破性进展将有助于LM的功能基因、毒力基因的研究。
突变株的生长、抗胁迫能力以及RealTime-PCR等实验结果表明,inlA和inlB基因的缺失对LM的生长和抗胁迫能力没有影响,但inlA和inlB基因的缺失造成了多个毒力基因的表达出现不同程度的变化。根据RealTime-PCR的结果可以初步推测,LM在缺失inlA和inlB基因后,由于hly、inlC等主要用于介导LM入侵宿主细胞以及prfA、sigB等调控基因的表达量下降,其毒力会明显下降,且主要体现在入侵宿主细胞能力的减弱,但由于两株突变株的actA基因表达没有明显变化,因此突变株在胞内寄生、运动以及胞间传播的能力可能没有变化。由于基因调控的复杂性和本实验中RealTime-PCR针对的毒力基因有一定的局限性, LM△inlA和LM△inlB毒力的具体变化情况,还需要进行后续的动物实验、细胞侵袭实验来验证。
[1] Kanki M,Naruse H,Taguchi M,et al.Characterization of specific alleles in InlA and PrfA ofListeriamonocytogenesisolated from foods in Osaka,Japan and their ability to invade Caco-2 cells[J].International Journal of Food Microbiology,2015,211:18-22.
[2] Travier L,Guadagnini S,Gouin E,et al.ActA PromotesListeriamonocytogenesAggregation,Intestinal Colonization and Carriage[J].Plos Pathogens,2013,9(1):430-445.
[3] Pilchová T,Hernould M,Prévost H,et al.Influence of food processing environments on structure initiation of static biofilm ofListeriamonocytogenes[J].Food Control,2014,35(1):366-372.
[4] Reniere M L,Whiteley A T,Hamilton K L,et al.Glutathione activates virulence gene expression of an intracellular pathogen[J].Nature,2015,517(7533):170-173.
[5] Bierne H,Sabet C,Personnic N,et al.Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes[J]. Microbes & Infection, 2007,9(10):1156-1166.
[6] 冯莹颖, 张强, 黄兰红,等. InlA和InlB介导单核细胞增生李斯特菌入侵宿主细胞分子机制的研究进展[J]. 微生物学通报, 2009, 36(22):1894-1900.
[7] Huang Y,Suo Y,Shi C,et al.Mutations ingltB andgltC reduce oxidative stress tolerance and biofilm formation inListeriamonocytogenes4b G[J].International Journal of Food Microbiology,2013,163(2-3):223-230.
[8] Suo Y,Huang Y,Liu Y,et al.The Expression of Superoxide Dismutase (SOD) and a Putative ABC Transporter Permease Is Inversely Correlated during Biofilm Formation inListeriamonocytogenes4b G[J].Plos One,2012,7(10):e48467-e48467.
[9] Park S F, Stewart G S A B.High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells[J].Gene,1990,94(1):129-132.
Construction ofinlA &inlB Genes Deletion Strain ofListeriamonocytogenes
LIU Wu-kang1, LI Sen1, CHEN Guo-wei1, LUO Qin2, WU Man1,DING Cheng-chao1, XIE Man-man1, LIU Qing1
(1.Schl.ofMed.Instrum't&FoodEngin.,Uni.ofShanghaiforSci. &Technol.,Shanghai200093;2.Coll.ofLifeSci.,CentralChinaNormalUniversity,Wuhan430079)
Listeriamonocytogenes(LM) is a food-borne pathogen shared by human being and animals which has a strong resistance to the adverse environment, and widely exists in various food processing environment and easily causes severe food safety problems. LM is a kind of intracellular parasitic bacterium, its virulence factor internalin A (InlA) and internalin B(InlB)are deemed to play important roles in LM penetrating the host barrier, invading the host cell and the transmission between cells. This paper adopted homologous recombination method to knock out theinlA andinlB genes of the wild strain EGDe and carried out the research on the basic biological characteristics ofinlA andinlB deletion strains, and applied RealTime-PCR to monitored the LM virulence genes’expression. The experimental results showed that its growth and tolerance of NaCl and ethanol (EtOH) were not affected by the mutation, but many virulence genes’expression was significantly decreased. The construction of deletion mutant strains provided important materials for further study on the specific function ofInlA andInlB in the process of LM invading to the host cell.
Listeriamonocytogenes; internalin A; internalin B; gene knock-out; RealTime-PCR
国家自然科学基金项目(31371776)
刘武康 男,硕士研究生。研究方向为食源性致病菌致病机理。E-mail:liu_wukang@126.com
* 通讯作者。男,博士,教授。研究方向为食源性致病菌致病机理及快速检测技术。E-mail:liuq@usst.edu.cn
2016-04-08;
2016-06-27
Q933
A
1005-7021(2017)01-0064-06
10.3969/j.issn.1005-7021.2017.01.010
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
睿士(2023年9期)2023-09-20 05:47:07
华人时刊(2023年1期)2023-03-14 06:43:36
云南化工(2021年6期)2021-12-21 07:31:04
汉字汉语研究(2021年2期)2021-08-30 08:58:46
农药科学与管理(2019年6期)2019-11-23 08:17:12
生物工程学报(2019年1期)2019-01-30 08:19:58
小雪花·初中高分作文(2016年9期)2016-05-14 02:50:08
河北书画研究(2016年3期)2016-04-28 08:55:35
华南农业大学学报(2015年5期)2015-12-04 03:04:38
