
Prokaryotic expression, purification and characterization of nattokinase
2017-04-27 07:49AIHaixinZHANGLiZHANGXingangCHENSiyaoHUHuanWEIHongyunMENGXinruiWANGTianqiZHAOJianLIUHongsheng
微生物学杂志 2017年1期
AI Hai-xin, ZHANG Li, ZHANG Xin-gang, CHEN Si-yao, HU Huan,3,WEI Hong-yun,4, MENG Xin-rui, WANG Tian-qi, ZHAO Jian, LIU Hong-sheng*
(1.School of Life Science,Liaoning University, Shenyang 110036, China;2.Research Center for Computer Simulating andInformation Processing of Bio-macromolecules of Liaoning, Shenyang 110036, China;3.Liaoning Engineering Laboratory for Molecular Simulation and Designing of Drug Molecules, Shenyang 110036, PR China;4.Sion Tiansheng Biological Technology Co.,Ltd.,Shenyang 110036,PR.China;5.School of Pharmacy, Liaoning University, Shenyang 110036, PR China)
Prokaryotic expression, purification and characterization of nattokinase
AI Hai-xin1,2,3, ZHANG Li1,2, ZHANG Xin-gang1, CHEN Si-yao1, HU Huan1,3,WEI Hong-yun1,4, MENG Xin-rui1, WANG Tian-qi5, ZHAO Jian1, LIU Hong-sheng1,2,3*
(1.School of Life Science,Liaoning University, Shenyang 110036, China;2.Research Center for Computer Simulating andInformation Processing of Bio-macromolecules of Liaoning, Shenyang 110036, China;3.Liaoning Engineering Laboratory for Molecular Simulation and Designing of Drug Molecules, Shenyang 110036, PR China;4.Sion Tiansheng Biological Technology Co.,Ltd.,Shenyang 110036,PR.China;5.School of Pharmacy, Liaoning University, Shenyang 110036, PR China)
Nattokinase is a fibrinolytic enzyme that is considered to be a promising agent for thrombosis therapy. In this study, nattokinase was purified from fermentation broth of aBacillussubtilisstrain by ammonium sulfate salting-out, gel filtration chromatography, and hydrophobic interaction chromatography with a purification fold of 5.2 and at a yield of 46.3%. The purified enzyme has molecular mass of 28 kDa and fibrinolytic activity of 4 580 U/mg. Since the concentration of nattokinase on fermentation broth was quite low, we cloned nattokinase gene fromB.subtilisand expressed it inE.coliBL21 (DE3). Nattokinase was actively expressed in the recombinant strain. The yield of nattokinase was increased significantly, but the activity of the protein produced by recombinant strain was low.
nattokinase;purification; prokaryotic expression
Nattokinase (NK, subtilisin NAT), a novel fibrinolytic enzyme, is one of the most considerable extracellular enzymes produced byBacillussubtilisin the traditional Japanese soybean food natto[1], the Chinese douchi[2], and other traditional fermented foods[3]. The enzyme has been proved to be a potent thrombolytic enzyme bothinvitroandinvivo[4-6]. Nattokinase could directly cleaves cross-linked fibrin, catalyzes the conversion of plasminogen to plasmin or inactivates the fibrinolysis inhibitor (PAI-1)invitro[7]. It is considered to be a promising natural agent for the prevention and treatment of embolism[3,4,7-9], since nattokinase have some advantages over other conventional fibrinolytic agents such as its food origin and relatively strong fibrinolytic activity. Microbial fibrinolytic enzymes for example nattokinase, have now attracted much more attention than typical thrombolytic agents which are more expensive and have some undesirable side effects[3].
Many studies have focused their efforts on the purification of nattokinase. In the process of purification, the traditional protein separation and purification techniques, such as organic solvent fractionation, salting out and protein chromatography, have been tested with a lot of disadvantages, such as long separation and purification time required, more operation units and less activity recovery rate[7,10]. In order to promote the purification process, some new technology have been applied to purify nattokinase from fermentation broth, for instance, reverse micelles extraction[11]and three phase partitioning[12]. In this study, we have aimed to purify nattokinase from fermentation broth ofBacillussubtilisby traditional purification techniques with high purification fold and high yield.
Since the concentration of nattokinase on fermentation broth is quit low, some efforts such as expression vectors with high structure stability, medium optimization, and promoter optimization have been made to enhance the production of nattokinase[13-16].Escherichiacoliis the most widely used prokaryotic system for heterologous protein production. Some studies have been attempt to express nattokinase inE.coli, but in most cases nattokinase was produced as inactive inclusion bodies[17-18], and could only be renatured protein from the inclusion bodies. Secretory expression of nattokinase inE.coliwas also reported in some recent study[19]on adding the signal peptide which could assist the secretion of expressed protein. In the present study, we have cloned and actively expressed the gene encoding nattokinase fromB.subtilisLNUB236 inE.coli.
1 Materials and Methods
1.1 Microorganism
The nattokinase producing strain,BacillussubtilisLNUB236, was isolated from natto by our group. The microorganism was maintained as spores suspended in 20% (v/v) glycerol, stored at -80 ℃.
1.2 Reagent and consumable
Sephadex G-75 was purchased from GE Healthcare Life Sciences (USA). Bovine fibrinogen and thrombin was obtained from National Instisutes for Food and Drug Control (China).TaqDNA Polymerase, T4DNA Ligase,EcoR I andBamH I were purchased from Fermentas Company (USA).PCR Purification kit,DNA gel extraction kit and plasmid extraction kit were purchased from Sion Tiansheng Biotech(China). DNA marker, low molecular weight protein standard, dNTP and IPTG were purchased from Sangon Biotech (China). Other common chemicals were of analytical reagent grade.
1.3 Fermentation
Nattokinase was produced byB.subtilisLNUB236 in a 30 L fermentor (Kobiotech, Korea). The seed culture was prepared in a 500 mL shark flask containing a 100 mL working volume of Luria-Bertani (LB)medium at 37 ℃ on a shaking incubator at 180 r/min for 12 h. The fermentor was inoculated with 800 mL of seed culture. The initial volume of the culture in fermentor was 20 L. The pH was controlled at a fixed value of 7.0. Temperature was controlled constant at 37 ℃. The dissolved oxygen (DO) level was automatically controlled at 20%. Agitation was performed at 130 r/min. The overall fermentation time was 48 hours.
1.4 Purification of nattokinase
All steps of the purification procedure were performed at 4 ℃. The fermentation broth was centrifuged at 8 000 r/min for 20 min. The supernatant was collected. Thirty percent of ammonium sulfate (w/v) was added to the supernatant with constant stirring in a magnetic stirrer. This fermentation-ammonium sulfate mixture stood overnight. The mixture was then centrifuged at 10 000 r/min for 30 min. More ammonium sulfate was added to the supernatant to increase the proportion of salt to 60%. This mixture stood overnight. The sedimentation was then collected and dissolved in phosphate buffer saline (PBS) after centrifugation of the mixture at 10 000 r/min for 30 min. The small molecule impurities and excess salt was removed by Labscale TFF System (Millipore, USA).
The enzyme dialysate was applied to a Sephadex G-75 column (1.0 cm ×150 cm) previously equilibrated with PBS. The column was eluted with PBS at a flow rate of 0.7 mL/min. Elution peaks were collected, and the enzyme activity in each fraction were determined. The fraction containing the enzyme was collected, and then loaded onto the Phenyl-Sepharose 6 Fast Flow column (1.6 cm×25 cm), which was previously equilibrated with PBS containing 1.0 mol/L ammonium sulfate. Proteins were eluted with a discontinuous gradient of ammonium sulfate ranging from 1.0 to 0 mol/L at a flow rate of 0.8 mL/min. The fractions were collected and determined by SDS-PAGE and fibrin plate assay.
1.5 Detection of nattokinase
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out to determine the purity and the molecular mass of purified enzyme. Protein bands were stained by Coomassie R 250 brilliant blue. Fibrinolytic activity was determined by measuring the areas of the lysed zone on the fibrinogen (bovine) plate. Ten microliter of the enzyme was dropped onto the small holes in the fibrin plate. The fibrin plate was then incubated for 18 h at 37 ℃。The fibrinolytic activity was estimated by measuring the diameter of the clear zone on the fibrin plate. Urokinase was used as a standard.
1.6 Expression nattokinase inE.coli
Total genomic DNA fromB.subtilisLNUB236 was extracted by Proteinase K method and then was used as a template for PCR amplification of the gene encoding the pro nattokinase. A pair of primers F-NK: 5′-GGCGGGATCCAGGGTAAAGAGTGAGAAGC-3′and R-NK: 5′-GCCGGAATTCGTCCAAGTCGGGTGC-3′ (the restriction sitesBamH I andEcoR I are underlined) were designed according to the DNA sequence of nattokinase precursor fromB.subtilisYF038 (GenBank accession number AY219901[20]). PCR was performed with initial denaturation at 94 ℃ for 5 min, followed by 30 cycles consisting denaturation at 94 ℃ for 30 s, annealing at 55 ℃ for 30 s, and extension at 72 ℃ for 30 s, and a finally extension at 72 ℃ for 7 min.Total volume of reactants was 50 μL.
The PCR products and the plasmid pET-26b(+) were double-digested byBamH I andEcoR I and then connected by T4-DNA ligase. The recombinant was transformed intoE.coliBL21 (DE3) competent cells and plated onto LB plates with 50 μg/mL kanamycin. The plasmid isolated from the positive transformant was sequenced. Andthe resulting sequence was then analyzed by NCBI BLAST (http://www.ncbi.nlm.nih.gov/BLAST/).
The selected transformants were cultured in LB plates containing 50 μg/mL kanamycin at 37 ℃. Then a positive colony was inoculated into a 50 mL liquid LB culture containing 50 μg/mL kanamycin. After incubation on a rotary shaker (37 ℃, 180 r/min) for 12 h, the culture was 1∶100 diluted into LB culture containing 50 μg/mL kanamycin and then shaken at 180 r/min and 37 ℃ for 3 h. The 1 mmol/L IPTG was added into LB culture for expressing the nattokinase gene. The culture was centrifuged for 20 min. The precipitation were lysed by sonication treatment. The fibrinolytic activity of the supernate was detected by fibrin plate method. The nattokinase expression level was detected by SDS-PAGE.
2 Result and discussion
2.1 Batch fermentation in a fermentor
The bacterial growth curve for the fermentation in a 30 L fermentor were determined by measuring the optical density (OD) of the culture at 600 nm with a spectrophotometer. As shown in Fig.1A, the bacterial growth curve was a typical sigmoidal shape (S-shape) and theOD600reached a maximum of 0.7 at 14 h. The fibrinolytic activity of the culture (nattokinase activity) was estimated by fibrin plate method. As shown in Fig.1B, the highest fibrinolytic activity of the culture reached 680 unit/mL at 24 h.
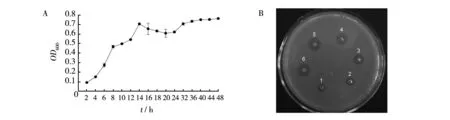
Fig.1 Analysis of nattokinase production by B. subtilis LNUB236 in a 30 L fermentorA: The growth curve;B: Fibrinolytic activity detection of the culture,1 to 6 indicate the fibrinolytic activity of the culture at 4, 8, 12, 24, 36,48 h, respectively
2.2 Purification of nattokinase
The procedure of purification nattokinase from the culture supernatant is described under ‘Materials and methods’. The supernatant was submitted to ammonium sulfate salting-out, gel filtration chromatography, and hydrophobic interaction chromatography. The yield and purity of the protease in each purification procedure were summarized in Table 1. The ammonium sulfate precipitation of the culture supernatant resulted in a 1.59-fold increase in the specific activity. The elution diagram of the Sephadex G-75 gel filtration (Fig.2A) showed three peaks. The fibrinolytic activity of each peak was tested by fibrin plate method (Fig.2B). The second peak showed highest specific activity. After the purification procedure on Sephadex G-75, a 2.9-fold activity increase was obtained. The eluant collected from peak 2 was submitted to the hydrophobic interaction chromatography. After the final purification on Phenyl-Sepharose 6 Fast Flow, a 5.2-fold purification and 46.3% yield were obtained.
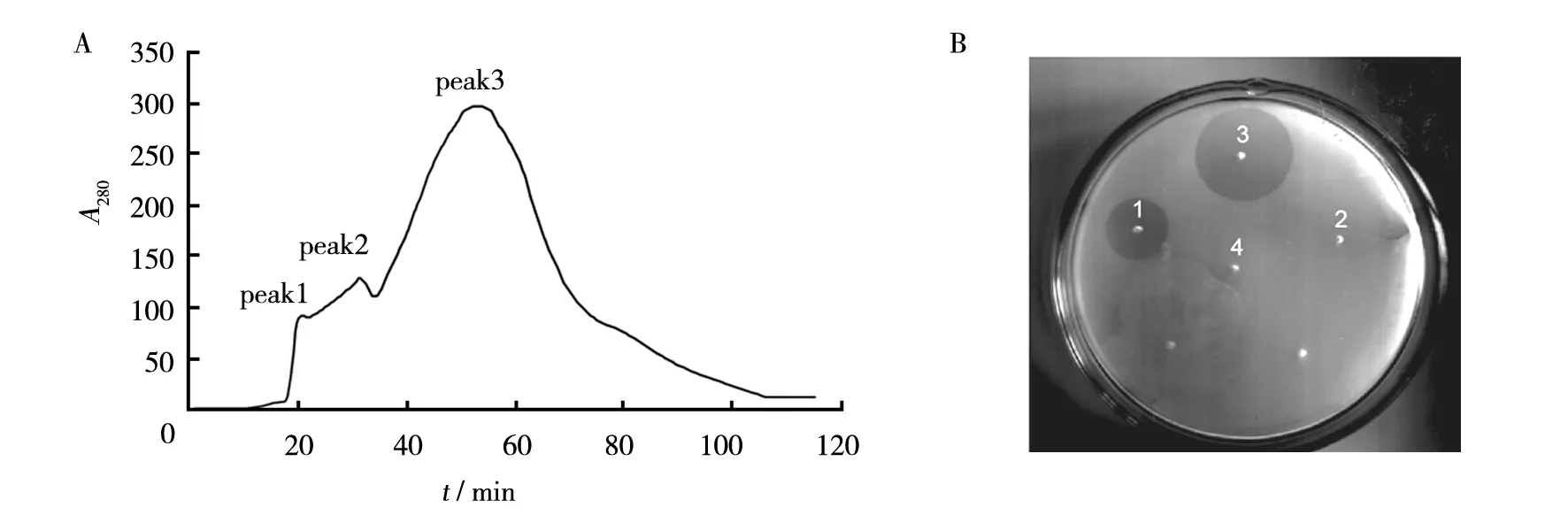
Fig.2 Purification of nattokinase by gel filtrationA: Elution diagram of the Sephadex G-75 gel filtration, Three peaks are shown in the diagram;B: The fibrinolytic activity of each peak. 1, 2, 3, and 4 indicates the activity of fermentation supernatant, peak 1, peak 2, and peak 3, respectively
After all of the steps, the purity of nattokinase on each process was determined by SDS-PAGE. After the step of Phenyl-Sepharose 6 Fast Flow, the enzyme was showed a single band with an apparent molecular mass of 29 kDa (Fig.3), indicating that the protein is purified.

Fig.3 SDS-PAGE analysis of the purified proteins from the various purification stepsM:molecular mass markers; 1:purified by Phenyl-Sepharose 6 Fast Flow; 2: purified by Sephadex G-75;3:culture supernatant
In the test for the effect of temperature on nattokinase, the purified nattokinase was dissolved in Tris-HCl (pH 7.0)buffer and placed at 20, 30, 40, 50, 60, and 70 ℃ for 30 min (Fig.4A). For the effect of pH, the enzyme solution was adjusted to pH 2.0,4.0,6.0,8.0,10.0, and 12.0, keptat 4 ℃ for 48 h (Fig.4B). Finally, the fibrinolytic activity of all samples was tested, and the results revealed that the purified nattokinase wasstable at the pH value of 8.0-10.0 and below 50 ℃. The enzyme shows considerable fibrinolytic activity at pH 6-11 but lacks functional and structural stability in alkaline environments. The enzyme activity also decreases sharply at temperatures beyond 60 ℃[7]. NK retains more than 95% of its activity after five rounds of freezing and thawing[1].
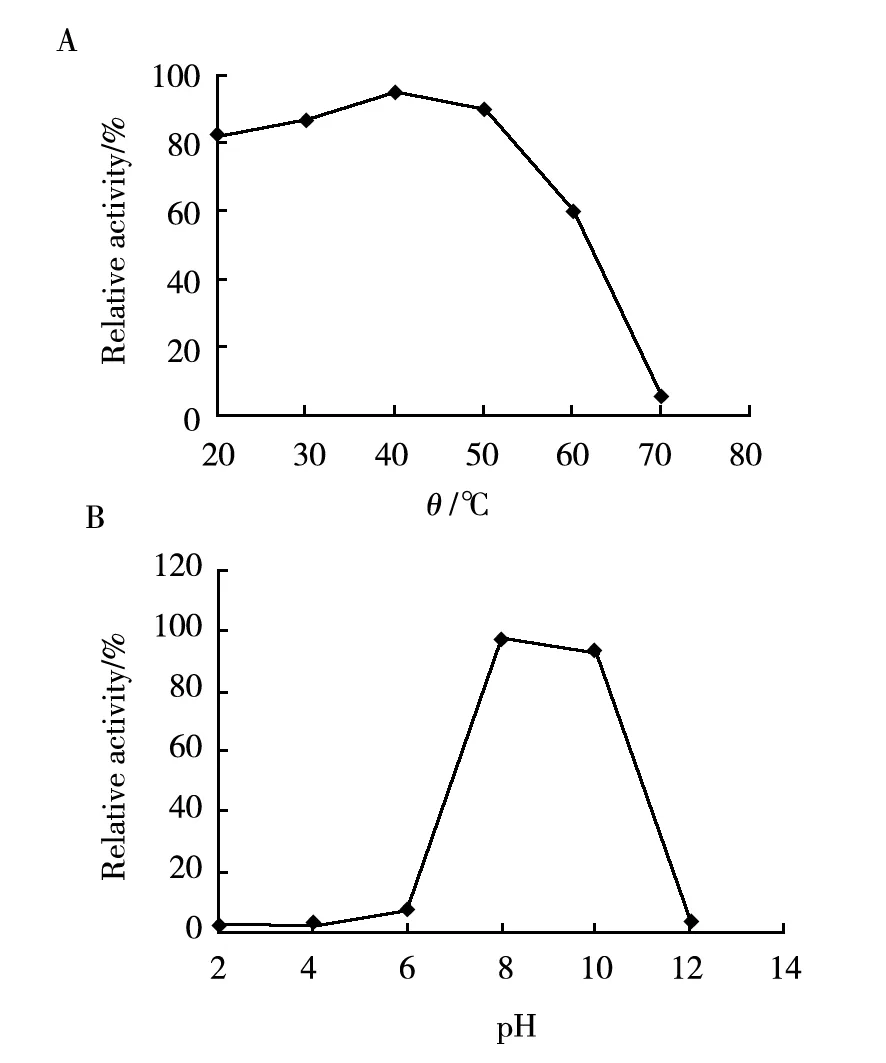
Fig.4 Effects of temperature (A) and pH (B) on the purified nattokinase
The fibrinolytic activity of the purified nattokinase kept under pH 7.0 at 4 ℃ and without inhibitorwas set as 100% standard, the relative activity was calculated bycomparison of the fibrinolytic activities under different conditions tothe 100% standard activity.
2.3 Cloning and expression of nattokinase inE.coli
DNA fragment encoding the pro nattokinase gene was amplified by PCR from the total DNA ofB.subtilisLNUB236. A single band of about 1 056 bp was obtained and sequenced (Fig.5). The sequence of the DNA fragment was almost identical to the DNA sequence of nattokinase precursor fromB.subtilisYF038 (GenBank accession number AY219901). Then the purified DNA fragment was ligated to the pET-26b(+) vector by T4DNA ligase. The ligation mixture was transformed intoE.coliBL21 (DE3) competent cells and plated onto LB plates. The recombinant strain was screened by LB plates containing 50 mg/mL of kanamycin. A positive clone was obtained by colony PCR.
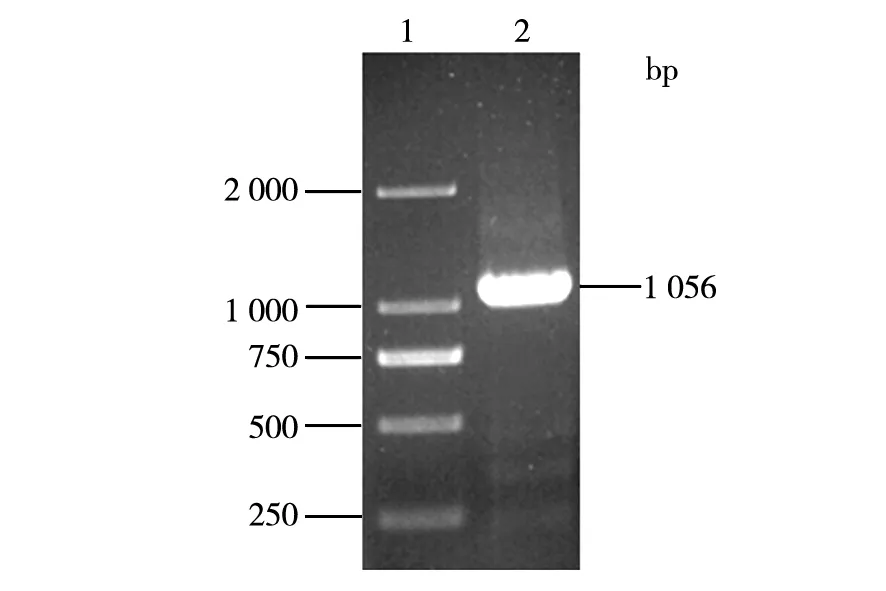
Fig.5 Amplification ofpro nattokinase gene1:DNA maker 2 000 bp;2:Pro nattokinase DNA fragment by PCR
After standard growth and induction of the positive transformant, the culture was subjected to the ultrasonication. Then the supernatant and precipitate were examined by SDS-PAGE. A major protein band of approximately 37 kDa was observed, which was identical to the molecular mass of nattokinase fromB.subtilis, while no counterpart band was observed in control strain (Fig.6A). The expression quantity of nattokinase was quite high in nutrient solution. By BandScan analysis, the amount of recombinant protein expression accounted for 42.9% of total protein on fermentation broth. The pro nattokinase (37 kDa) was predominately found in the soluble fraction of cell lysate after centrifugation, but its enzymatic activity was very low (Fig.6B).
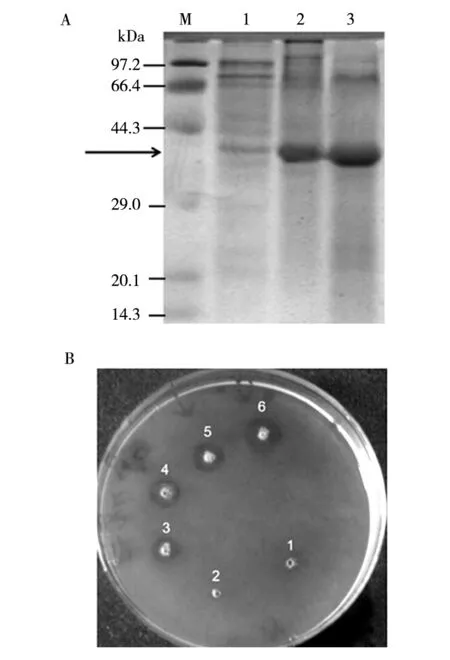
Fig.6 Detection of the expression and the activity of nattokinase in E. coli by SDS-PAGE and fibrin plate A: SDS-PAGE showing that the 37 kDa pro nattokinase (indicated by arrow) was expressed in the recombinant E. coli BL21 (DE3),M: molecular weight markers, 1: fermentation broth without containing IPTG, 2: the supernatant after ultrasonication induced by IPTG,3: the precipitate after ultrasonication induced by IPTG;B: Fibrinolytic activity assay results of expressed products,1 and 2 are the fermentation broth without containing IPTG, 3 and 4 are the precipitate after ultrasonication, 5 and 6 are the supernatant after sonic disruption
The activity of the supernatant was only 6.1 IU/mL, but that of the precipitate was 21.7 IU/mL. Furthermore, One of the target fragment only contained mature peptide of nattokinase gene was expressed in the pET-26b(+) vector. But neither the supernatant nor the precipitate of the fermented culture of the vector carried the mature nattokinase gene showed fibrinolytic activity in the fibrin plate assays. The propeptide has been proved to function as an intramolecular chaperone and is essential for guiding the correct folding of the linked protein to its mature form[21-22]. The propeptide of subtilisin[23]was also reported to be necessary for active expression.
3 Conclusions
In the present study, we purified nattokinase from the supernatant ofB.subtilisLNUB236 fermentation broth in a 30 L fermentor by ammonium sulfate salting-out, gel filtration chromatography, and hydrophobic interaction chromatography. After a series of purification, the purification fold of nattokinase was improved from 1 to 5.2, the yield declined from 100% to 46.7%. Compared with other purification methods, the purity was low, but the yield was quite high. Therefore, further studies could be focused on the improvement of the purification fold. Since the concentration of nattokinase on fermentation broth was quit low, we cloned the DNA fragment encoding pro nattokinase fromB.subtilisLNUB236 and expressed it inE.ColiBL21. The protein production increased significantly, the amount of recombinant protein expression accounted for 42.9% of total protein on fermentation broth, but the activity of the protein produced by recombinant strain was quite low. Therefore, the further study is to construct the new recombinant strain that can produce nattokinase with high activity.
Reference
[1] Sumi H, Hamada H, Tsushima H, et al. A novel fibrinolytic enzyme (nattokinase) in the vegetable cheese Natto; a typical and popular soybean food in the Japanese diet[J].Experientia, 1987,43(10):1110-1111.
[2] Peng Y, Yang X J, Xiao L, et al. Cloning and expression of a fibrinolytic enzyme (subtilisin DFE) gene fromBacillusamyloliquefaciens DC-4 inBacillussubtilis[J].Research in microbiology, 2004,155(3): 167-173.
[3] Peng Y, Yang X, Zhang Y. Microbial fibrinolytic enzymes: an overview of source, production, properties, and thrombolytic activityinvivo[J].Applied microbiology and biotechnology, 2005,69(2):126-132.
[4]Fujita M, Hong K, Ito Y, et al. Transport of nattokinase across the rat intestinal tract[J].Biological & pharmaceutical bulletin, 1995,18(9):1194-1196.
[5] Xu J, Du M, Yang X, et al. Thrombolytic effectsinvivoof nattokinase in a carrageenan-induced rat model of thrombosis[J].Acta haematologica, 2014,132(2): 247-253.
[6] Unrean P, Nguyen N H. Metabolic pathway analysis and kinetic studies for production of nattokinase inBacillussubtilis[J].Bioprocess and biosystems engineering, 2013,36(1): 45-56.
[7] Fujita M, Nomura K, Hong K, et al. Purification and characterization of a strong fibrinolytic enzyme (nattokinase) in the vegetable cheese natto, a popular soybean fermented food in Japan[J].Biochemical and biophysical research communications, 1993,197(3):1340-1347.
[8] Sumi H, Hamada H, Nakanishi K, et al. Enhancement of the fibrinolytic activity in plasma by oral administration of nattokinases[J].Acta haematologica, 1990,84(3):139-143.
[9] Dabbagh F, Negahdaripour M, Berenjian A,et al. Nattokinase: production and application[J].Applied microbiology and biotechnology, 2014,98(22): 9199-9206.
[10]Urano T, Ihara H, Umemura K, et al. The profibrinolytic enzyme subtilisin NAT purified fromBacillussubtiliscleaves and inactivates plasminogen activator inhibitor type 1[J].Journal of Biological Chemistry, 2001,276(27): 24690-24696.
[11]Liu J G, Xing J M, Chang T S, et al. Purification of nattokinase by reverse micelles extraction from fermentation broth: effect of temperature and phase volume ratio[J].Bioprocess and biosystems engineering, 2006, 28(4):267-273.
[12]Garg R, Thorat B N. Nattokinase purification by three phase partitioning and impact of t-butanol on freeze drying[J].Separation and Purification Technology, 2014,131(2):19-26.
[13]Mahajan P M, Gokhale S V, Lele S S. Production of nattokinase usingBacillusnatto NRRL 3666: Media optimization, scale up, and kinetic modeling[J].Food Science and Biotechnology, 2010,19(6):1593-1603.
[14]Wu S M, Feng C, Zhong J, et al. Enhanced production of recombinant nattokinase inBacillussubtilisby promoter optimization[J].World Journal of Microbiology and Biotechnology, 2011,27(1):99-106.
[15]Wang J K, Chiu H H, Hsieh C S. Optimization of the medium components by statistical experimental methods to enhance Nattokinase activity[J].Fooyin Journal of Health Sciences, 2009,1(1): 21-27.
[16]Chen P T, Chao Y P. Enhanced production of recombinant nattokinase inBacillussubtilisby the elimination of limiting factors[J].Biotechnology letters, 2006,28(19):1595-1600.
[17]Zhang R H, Xiao L, Peng Y, et al. Gene expression and characteristics of a novel fibrinolytic enzyme (subtilisin DFE) inEscherichiacoli[J].Letters in applied microbiology, 2005,41(2):190-195.
[18]Choi N S, Chang K T, Jae Maeng P, et al. Cloning, expression, and fibrin (ogen) olytic properties of a subtilisin DJ-4 gene fromBacillussp.DJ-4[J].FEMS microbiology letters, 2004,236(2): 325-331.
[19]Liang X, Jia S, Sun Y, et al. Secretory expression of nattokinase fromBacillussubtilisYF38 inEscherichiacoli[J].Molecular biotechnology, 2007,37(3):187-194.
[20]Nakamura T, Yamagata Y, Ichishima E. Nucleotide sequence of the subtilisin NAT gene, aprN, ofBacillussubtilis(natto)[J].Bioscience, biotechnology, and biochemistry, 1992,56(11): 1869-1871.
[21]Ikemura H, Takagi H, Inouye M. Requirement of pro-sequence for the production of active subtilisin E inEscherichiacoli[J].Journal of Biological Chemistry, 1987,262(16):7859-7864.
[22]O’Donohue M J, Beaumont A. The roles of the prosequence of thermolysin in enzyme inhibition and foldinginvitro[J].Journal of Biological Chemistry, 1996, 271(43): 26477-26481.
[23]Fu X, Inouye M, Shinde U. Folding pathway mediated by an intramolecular chaperone the inhibitory and charperone functions of the subtilisin propeptide are not obligatarily linked[J].Journal of Biological Chemistry, 2000, 275(22):16871-16878.
辽宁省科技厅基金资助项目(2013225086,2014001015);辽宁省教育厅基金资助项目(L2014001,LT2015011);
艾海新 男,博士,副教授。主要研究方向为分子微生物学。E-mail:ahxmail@163.com
纳豆激酶的原核表达、纯化及其特性分析
艾海新1,2,3, 张 力1,2, 张新刚1, 陈思瑶1, 胡 桓1,3,魏洪运1,4, 蒙新睿1, 王天琦5, 赵 健1, 刘宏生1,2,3*
(1.辽宁大学 生命科学院,辽宁 沈阳 110036;2.辽宁省生物大分子计算模拟与信息处理工程技术研究中心,辽宁 沈阳 110036;3.辽宁省药物分子模拟与设计工程实验室,辽宁 沈阳 110036;4.沈阳神州天盛生物科技有限公司,辽宁 沈阳 110036;5.辽宁大学 药学院,辽宁 沈阳 110036)
纳豆激酶 (Nattokinase)作为一种新型溶解血栓的纤维蛋白溶解酶,有着广阔的市场前景和巨大的产业化潜力。本研究从枯草芽胞杆菌(Bacillussubtilisnatto)发酵液中提取、纯化纳豆激酶,依次通过硫酸铵盐析、凝胶过滤层析和疏水层析方法纯化纳豆激酶,纯化倍数为5.2,纯化率为46.3%。纯化后的纳豆激酶经SDS-PAGE电泳显示相对分子量约为28 kDa,纤维蛋白平板法显示活性为4 580 IU/mg。但纳豆激酶在枯草芽胞杆菌发酵液中的含量较低,因此在原核表达载体大肠埃希菌(Escherichiacoli)BL21(DE3)中克隆并高效表达了纳豆激酶基因,其纳豆激酶产量显著提高,但酶活相对降低。
纳豆激酶;纯化;原核表达
Q939.9-3
A
1005-7021(2017)01-0020-08
沈阳市科技局基金资助项目(F15165400,F16205151)
*通讯作者。男,博士,教授。主要研究方向为分子微生物学。 Tel:024-62202280,E-mail:liuhongsheng@lnu.edu.cn
10.3969/j.issn.1005-7021.2017.01.004
Funds Project: support by Nature Science Foundation (2013225086);Science of Public Research Foundation (2014001015) from the
Scientific and Technological Department of Liaoning Province and Innovation Team Project (LT2015011);General Research Project
Foundation (L2014001) from the Education Department of Liaoning Province;Large-scale Equipment Shared Services Project
(F15165400);Applied Basic Research Project (F16205151) from the Science and Technology Bureau of Shenyang
Author profile: Ai Haixin male, PhD, Associate Professor.Research interests:molecular microbiology.E-mail:ahxmail@163.com
*Corresponding author:male,PhD, Professor. Research interests:molecular microbiology. Tel:024-62202280,
E-mail:liuhongsheng@lnu.edu.cn
Draft accepted date:2016-03-18; Revision returned date:2016-04-01
猜你喜欢
中国动物保健(2022年2期)2022-05-05
新生代(2019年4期)2019-11-13
现代营销(创富信息版)(2018年5期)2018-07-12
现代营销(创富信息版)(2018年4期)2018-06-01
现代营销(创富信息版)(2018年3期)2018-03-15
现代营销(创富信息版)(2018年2期)2018-02-10
辽宁大学学报(自然科学版)(2017年3期)2017-11-24
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
安徽医科大学学报(2015年9期)2015-12-16
现代检验医学杂志(2015年5期)2015-02-06
