
新生儿非酮症性高甘氨酸血症1例
2017-04-08 04:43:31戴红梅蔡蕾伍志翔
神经损伤与功能重建 2017年2期
戴红梅,蔡蕾,伍志翔
新生儿非酮症性高甘氨酸血症1例
戴红梅,蔡蕾,伍志翔
新生儿;非酮症性高甘氨酸血症;基因突变;个案
1 临床资料
患儿,男,3 d,因“反应差3 d,呼吸衰竭14 h,抽搐1次”入我院。外院病史:患儿系G1P1,胎龄40+2周,平产出生,出生体重3200 g。生后出现进行性反应差、哭声弱。生后第2天就诊于当地医院新生儿科,体查:精神反应差,不哭,前囟平软;呼吸急促,双肺呼吸音低;四肢凉,张力不高,原始反射减弱。血气分析、血常规、血生化、脑脊液检查未见异常;胸片示新生儿肺炎;头颅CT示脑白质密度减低,见图1。因不能排除感染,该院予头孢哌酮舒巴坦输液治疗,但患儿气促进行性加重,吸氧不能缓解。于生后第3天开始出现呼吸暂停、发绀、氧饱和度下降,并出现抽搐1次,为双下肢抖动,持续约 5 min,当地医院予苯巴比妥钠静脉滴注止痉后由当地120转送至我院。入院后(即生后第4天)体查:神志昏迷,弹足底无反应、不哭。全身苍白,皮肤巩膜中度黄染,四肢凉,毛细血管充盈时间延长。双侧瞳孔对光反射迟钝,无自主呼吸。振幅整合脑电图(aEEG)检查示:以不连续图形为主,少量交替图形,可见少量中低波幅尖波、棘波散发,脑电活动轻度异常,见图2。入院后改用“美罗培南”加强抗感染、呼吸机辅助通气及营养支持治疗后病情无好转,仍持续昏迷。家属考虑到预后差签字放弃治疗,患儿出院后死亡。5 d后血氨基酸及酯酰基肉碱谱分析(GC/MS)结果显示:血甘氨酸为 1304.32 mol/L(正常值130~650 mol/L)。遗憾的是此时家属已放弃治疗,未能进一步完善脑脊液的甘氨酸检测。但进一步用对其父母进行基因检测结果显示:其父母的甘氨酸脱羧酶基因(Glycine decarboxylase gene,GLDC)18号外显子上均有一个新的错义突变:c.2198 C>T(p.Ala733Val)。该基因突变在人类突变数据库尚未见记载,经保守性分析,该区为高度保守区域,怀疑是致病突变,见图4。
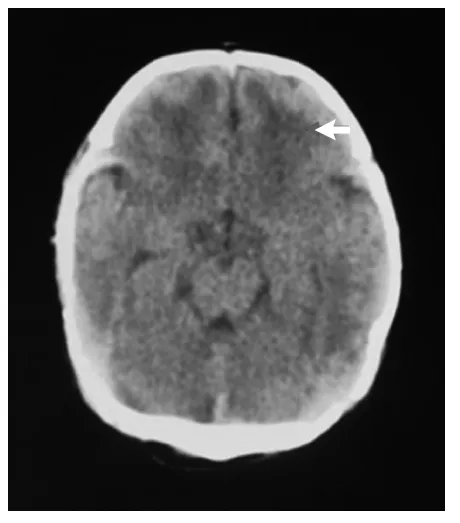
图1 患儿头颅CT平扫:脑白质密度减低(箭头所示)
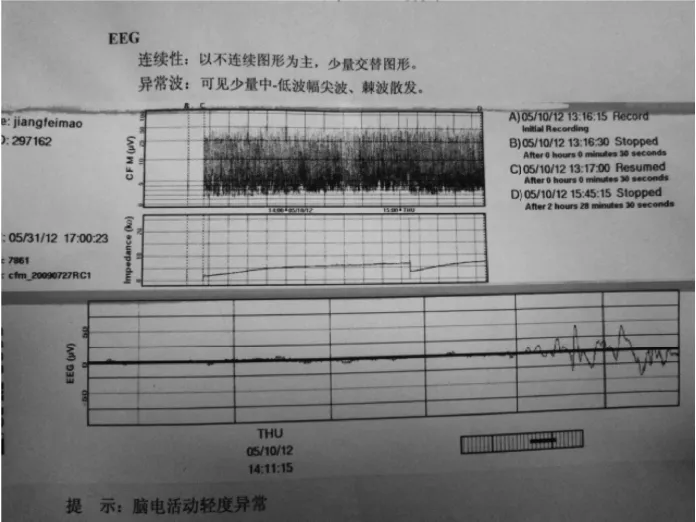
图2 患儿振幅整合脑电图
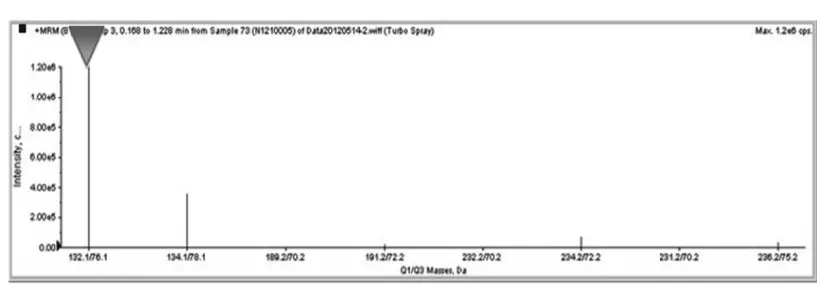
图3 血氨基酸串联质谱图
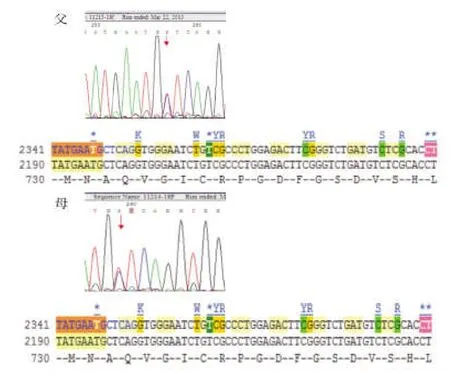
图4 患儿父母甘氨酸脱羧酶基因(GLDC)测序结果
2 讨论
非酮症性高甘氨酸血症(nonketotic hyperglycemia,NKH)是一种累及神经系统发育的常染色体隐性遗传病,有文献通过关于55 000例新生儿的流行病学调查发现其中NKH的发病率约为1/63 000[1]。尽管NKH发病率很低,但我国每年出生人口众多,应该有相当数量的NKH病例,但迄今为止也只有个例文献报道[3]。1969年Tada[2]等率先发现该病并指出NKH的发病机制是由于甘氨酸裂解系统(glycine cleavage system,GCS)活性降低所致。甘氨酸在体内的合成代谢中具有重要的生理作用,主要通过GCS进行。GCS活性降低可导致甘氨酸在体内异常堆积,造成神经系统发育障碍和脑功能受损。甘氨酸在中枢神经系统通过2种受体发挥作用:一种是抑制性受体,分布在脑干和脊髓,患者可出现呼吸暂停,肌张力减低,呃逆;另一种则是兴奋性受体,在大脑皮质和前脑等部位,作用于N-甲基-D-天冬氨酸受体(N-methyl-D-aspartic acid receptor,NMDAR),患者则出现癫痫、抽搐、不自主舞蹈。该病的临床表现不一,可分为3种类型:新生儿型、非典型性和暂时型。新生儿型又称为经典型,最常见,约占84%。该类疾病患儿出生6 h~8 d内出现进行性加重的嗜睡、肌张力减低、喂养困难和呃逆,进而出现呼吸衰竭,若无呼吸机辅助通气通常会导致昏迷和死亡。本型患儿约30%在新生儿期死亡,即便存活也多数出现严重的神经系统运动障碍,如持续癫痫发作,运动和认知障碍[8]。
NKH患者的临床表现通常不典型,生化检查如电解质测定、血气、尿液有机酸均可正常,无酮症酸血症;最初的EEG通常呈现爆发抑制表现为节律紊乱或多灶性棘波;MRI可正常或显示胼胝体发育不全,脑白质密度减低;确诊需依赖测定血浆与脑脊液中甘氨酸水平,甘氨酸在脑脊液/血浆>0.08是诊断NKH的重要依据;进行肝穿刺活检检查GCS活性以明确是何种蛋白缺失或者进行基因检测,发现GCS基因突变可明确诊断;Kure S[9]报道了两种新的诊断方法将有助于诊断NKH:[1-(13)C]甘氨酸呼气试验和多重连接依赖性探针扩增(MLPA)检测大片段缺失的GLDC基因。
编码P蛋白有关的GLDC基因缺失是导致NKH的主要原因[4]。至少有多于150种GLDC基因的错义突变与高甘氨酸血症相关,但是目前已经报道的关于GLDC基因的突变屈指可数。Yoon等[5]分别在GLDC基因的8号和18号外显子上发现错义突变 c.1130C>T(p.A377V)及 c.2081_2088del(p.A694DfsX11)。Love等[6]在GLDC基因19号外显子上发现错义突变c.2296G>T(p.Gly766Cys)。Brunel-Guitton等[7]报道了1例晚发型GDLC基因错义突变c.605C>T(p.Ala202Val),这是晚发型患者具有潜在遗传缺陷的第1次报告。本例患者血浆甘氨酸水平显著增高,且其父母基因检测均有相同位点的突变c.2198 C> T(p.Ala733Val)的错义突变,目前尚无文献报道。
该病目前尚无有效治疗方法,主要治疗药物有苯甲酸钠、氯胺酮和右美沙芬。苯甲酸钠与甘氨酸结合形成马尿酸盐从尿中排泄,可降低血甘氨酸水平[10];NMDA受体拮抗剂氯胺酮和右美沙芬对患儿的抽搐,癫痫及脑电图均有改善[11]。这些药物可改善患者的临床症状,但存活的患儿依旧有严重的神经系统后遗症,故有效的治疗仍需进一步研究。NKH患儿中新生儿型预后差,产前诊断尤为重要,本例患者的父母在下一次妊娠时可以通过胎盘绒毛GCS活性检测或者羊水中胎儿基因检测以确诊。
本例报告首次发现了GDLC基因的18号外显子上一个新的错义突变:c.2198 C>T(p.Ala733Val)。NKH的早期诊断很有挑战,因该病早期症状和体征都无显著特异性。若新生儿生后不久出现肌张力降低、嗜睡、呼吸暂停、喂养困难、呃逆或抽搐发作时,应考虑该病。基因检测目前已成为确诊NKH的重要手段,该病目前虽无特效治疗方法,即使早期诊断也不能逆转疾病神经功能结局,但可提供有效地遗传咨询及产前诊断,以降低该病的发病率。
[1]Applegarth DA,Toone JR,Lowry RB.Incidence of inborn errors of metabolism in British Columbia,1969-1996[J].Pediatrics,2000,105:e10.
[2]Tada K,Narisawa K,Yoshida T,et al.Hyperglycinemia:a defect in glycine cleavage reaction[J].Tohoku J Exp Med,1969,98:289-296.
[3]张蓉,陈超,曹云,等.新生儿非酮症性高甘氨酸血症1例[J].中国循证儿科杂志,2009,4:153-155.
[4]Kikuchi G,Motokawa Y,Yoshida T,et al.Glycine cleavage system:reaction mechanism,physiological significance,and hyperglycinemia[J]. Proc Jpn Acad Ser B Phys Biol Sci,2008,84:246-263.
[5]Yoon IA,Lee NM,Yoo BH,et al.Two novel missense mutations observed in nonketotic hyperglycinemia[J].Pediatr Neurol,2012,46: 401-403.
[6]Love JM,Prosser D,Love DR,et al.A novel glycine decarboxylase gene mutation in an Indian family with nonketotic hyperglycinemia[J].J Child Neurol,2014,29:122-127.
[7]Brunel-Guitton C,Casey B,Coulter-Mackie M,et al.Late-onset nonketotic hyperglycinemia caused by a novel homozygous missense mutation in the GLDC gene[J].Mol Genet Metab,2011,103:193-196.
[8]Suzuki Y,Kure S,Oota M,et al.Nonketotic hyperglycinemia:proposal of a diagnostic and treatment strategy[J].Pediatr Neurol,2010,43: 221-224.
[9]Kure S.Two novel laboratory tests facilitating diagnosis of glycine encephalopathy(nonketotic hyperglycinemia)[J].Brain Dev,2011,33: 753-757.
[10]Van Hove JL,Vande KK,Hennermann JB,et al.Benzoate treatment and the glycine index in nonketotic hyperglycinaemia[J].J Inherit Metab Dis,2005,28:651-663.
[11]Korman SH,Wexler ID,Gutman A,et al.Treatment from birth of nonketotic hyperglycinemia due to a novel GLDC mutation[J].Ann Neurol,2006,59:411-415.
(本文编辑:唐颖馨)
R741;R725
ADOI10.16780/j.cnki.sjssgncj.2017.02.033
中南大学湘雅三医院儿科长沙 410013
2016-09-21
戴红梅daihm1982@qq. com
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
上海计量测试(2022年4期)2022-02-01 07:41:18
中国现代医药杂志(2020年10期)2020-12-14 07:20:00
实用临床医学(2016年8期)2016-06-08 06:10:06
中国民族医药杂志(2016年2期)2016-05-14 07:11:57
人间(2015年11期)2016-01-09 13:12:58
邵阳学院学报(自然科学版)(2015年2期)2015-06-05 12:22:39
中国洗涤用品工业(2015年7期)2015-02-28 19:02:41
