
(S)-3-羟基四氢呋喃的合成研究进展
2017-04-01 02:52钟为慧
合成化学 2017年3期
郑 亿, 王 辉, 亓 亮, 凌 飞, 钟为慧
(浙江工业大学 药学院 长三角绿色制药协同创新中心,浙江 杭州 310014)
·综合评述·
(S)-3-羟基四氢呋喃的合成研究进展
郑 亿, 王 辉, 亓 亮, 凌 飞, 钟为慧*
(浙江工业大学 药学院 长三角绿色制药协同创新中心,浙江 杭州 310014)
综述了(S)-3-羟基四氢呋喃制备方法的研究现状,重点阐述了三种合成方法:(1)手性底物合成法;(2)手性催化剂不对称合成法;(3)酶催化不对称合成法。分析了不同制备方法的优劣,并对其未来发展进行了展望。参考文献45篇。
(S)-3-羟基四氢呋喃; 合成; 中间体; 综述
自上世纪60年代“反应停”事件后,美国FDA要求对外消旋药物进行拆分,合成具有光学活性的药物逐步成为手性药物研究的重要目标和手段[1-3]。
(S)-3-羟基四氢呋喃(1),是一种重要的医药化工中间体,在抗癌药、降糖药、艾滋病药等药物中用途广泛[4-10],例如可用于合成抗心律失常药物替卡地松(2),降糖类药物恩格列净(3),抗艾滋病药物安瑞那韦(4),抗癌药物阿法替尼(5)等(Chart 1)。目前1的市场售价为360 万·吨-1,仅国内就有145家制造商或贸易商,具有较高的市场价值。相对于(R)-3-羟基四氢呋喃而言,很多药物都含有1,且价格较低,因此往往将1作为中间体;而直接采用3-羟基四氢呋喃外消旋体来制备手性药物及中间体,后期拆分过程成本较高,不利于工业化生产。因此,在工业生产中1以其价格、手性等独特的优势而得到重视。本文根据近年来合成1的相关文献,系统地介绍了国内外的合成方法,并对各方法的优缺点进行简要评述。
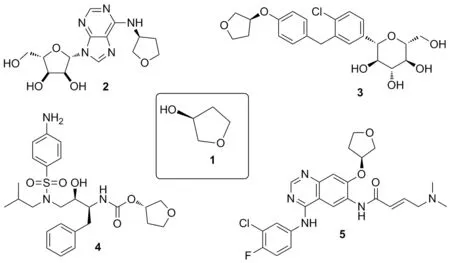
Chart 1
Scheme 1

Scheme 2
1 手性底物合成法
1.1 L-苹果酸(14)及衍生物为原料
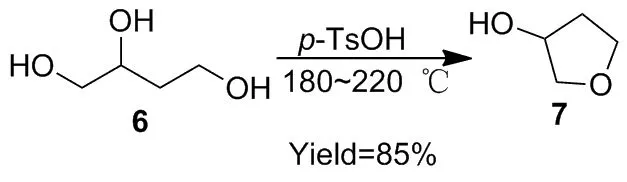
早在1958年,Wynberg[11-12]报道了以14的还原产物1,2,4-丁三醇(6)为原料,在一水合对甲苯磺酸催化下得到消旋体3-羟基四氢呋喃(7),收率85%(Scheme 1)。该方法无需溶剂,后处理直接分馏提纯,为合成1奠定了基础。
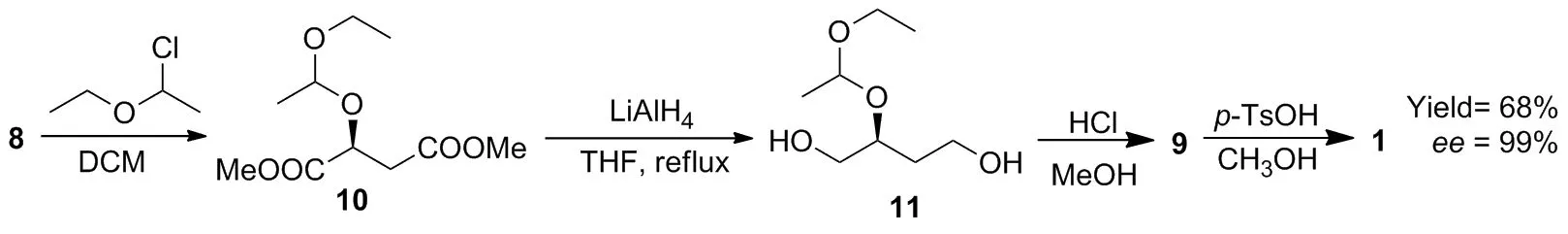
1983年,Tandon等[13-15]报道了以L-苹果酸二甲酯(8)为起始原料,用LiAlH4还原得到 (S)-1,2,4-丁三醇(9);中间体9在对甲苯磺酸催化下发生脱水环合反应得到产物1(Scheme 2),总收率为61%,ee值为94%。该路线具有原料价廉易得,步骤较短等优点,但8还原成中间体9时易发生外消旋化,且因中间体9水溶性较好,难以分离提纯。
为了减少外消旋化,Tandon等[16]对原有工艺进行改进,用烷基化试剂1-氯-1-乙氧基乙烷将8的2位羟基进行保护得到 (S)-2-(1′-(乙氧基)-乙氧基)-1,4-丁酸二甲酯(10),再用LiAlH4还原酯基得到 (S)-2-(1′-(乙氧基)-乙氧基)-1,4-丁二醇(11),随后脱保护得到9,最后环合得到目标产物1(Scheme 3)。改进后的路线有效地减少了外消旋化,总收率为68%,ee值达99%。但LiAlH4价格相对较贵,导致原料成本偏高,且中间体9难以提纯的问题依旧没有解决,不利于工业化应用。
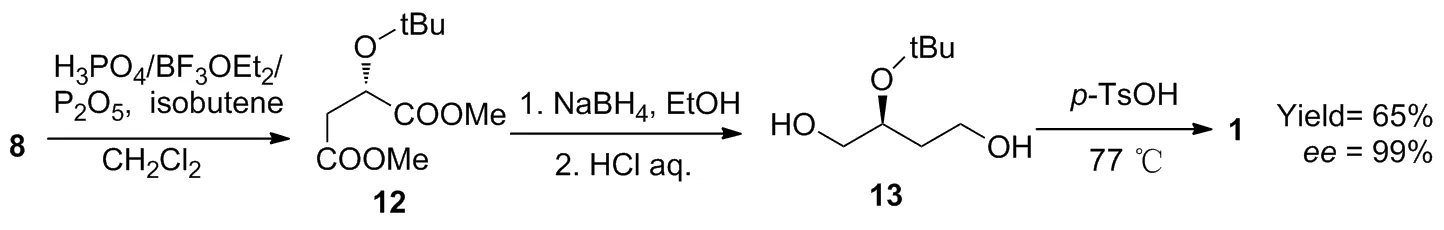
基于1广阔的应用前景,人们积极探索一条适合工业化生产的合成路线。2004年Izawa等[17-18]以L-苹果酸二甲酯8为原料,先用异丁烯将其2位羟基进行保护得到 (S)-2-叔丁氧基丁酸二甲酯(12);接着用廉价的NaBH4替代LiAlH4作还原剂,将12还原成 (S)-2-叔丁氧基丁三醇(13);最后在对甲苯磺酸催化下,13经2位脱保护、脱水环合反应,得到产物1(Scheme 4),收率为65%,ee值为99%。该路线用价廉易得的NaBH4作还原剂,大大降低了原料成本;对8先保护后还原,不仅从源头上避免了外消旋化,而且巧妙地避免了中间体8的2位羟基与硼试剂发生络合,还原产物13易分离提纯。
2007年,郑剑峰等[19-20]以14为原料,经酯化,还原,环合三步反应得到产物1,总收率为40%(Scheme 5)。该路线用LiCl/KBH4替代氢化铝锂进行还原反应,后处理采用无机酸,无需索氏提取器,不足之处是收率相对较低。
2008年顾松林等[21]同样以14为原料合成产物1,收率为76%(Scheme 6)。值得指出的是,该路线用无机酸和二氯亚砜共催化酯化反应,极大地减少了二氯亚砜的用量,减少了环境污染,此外,采用ZnCl2/NaBH4体系替代LiAlH4,提高了还原反应的收率。
Scheme 3
Scheme 4

Scheme 5
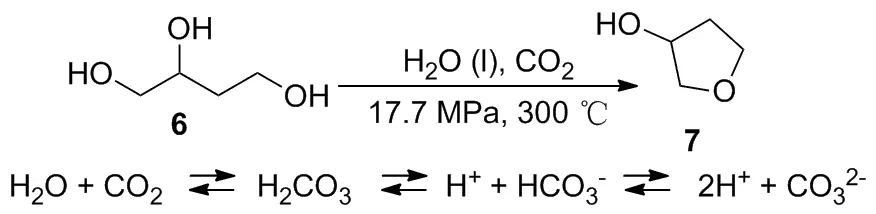
在1的制备中,脱水环合反应大多采用对甲苯磺酸作催化剂,副反应较多,收率低。2009年Shirai等[22]报道在高温高压的水和CO2介质中,6发生脱水环合反应得到7,收率70%(Scheme 7)。可能的反应机理是高温高压下,H2O与CO2反应生成H2CO3,后者解离出H+促进了脱水环合反应。我们推测若将底物6替换成9,也许可以得到1。该路线绿色无污染,但反应条件较为苛刻。
2012年,Rokicki等[23]报道在80 ℃下,以6为底物,K2CO3催化下与碳酸二甲酯(DMC)反应一步得到化合物7,收率高达93%(Scheme 8)。该路线试剂绿色环保,价廉易得,且反应后处理简单方便。同样的,若在6的2位引入手性,有可能高效地合成1,具有潜在的工业化前景。值得注意的是,DMC不宜过量,否则会生成副产物,影响反应收率和纯度,可能机理如Scheme 8所示。
1.2 (S)-4-氯-3-羟基丁酸乙酯(19)为原料
19是合成1的重要原料,可由4-氯-3-丁酮酸乙酯立体选择性还原得到[24-27]。在甲醇和四氢呋喃的混合溶剂中,于100 ℃, H2(1.5 MPa)氛围中,以[(R)-P-Phos-Ru(Ph)Cl]Cl为催化剂,实现羰基的不对称还原,得到99%的收率和98%的ee值(Scheme 9)。
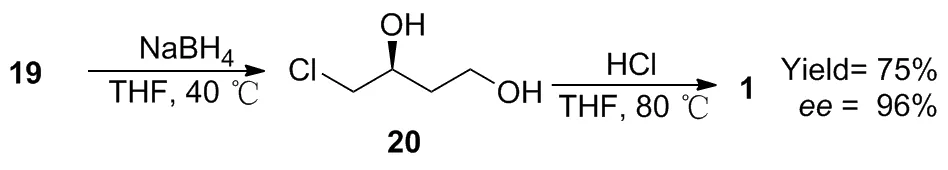
2008年,李勇智等[28]以19为原料,经NaBH4还原得到 (S)-4-氯-1,3-丁二醇(20),接着在酸性条件下80 ℃脱水环合得到1,产物总收率为75%,ee值为96%(Scheme 10)。该反应路线步骤短,收率较高,不足之处是还原反应易发生外消旋化而降低ee值。
Scheme 7

Scheme 8

Scheme 9
Scheme 10

Scheme 11

Scheme 12

Scheme 13
Scheme 14
2010年,陆军等[29]报道以19为起始原料,以异丁烯保护19的2-位羟基得到 (S)-4-氯-3-叔丁氧基丁酸乙酯(21),再经NaBH4还原得到 (S)-4-氯-3-叔丁氧基丁醇(22),接着在碱性条件下环合得到 (S)-3-叔丁氧基四氢呋喃(23),最后通HCl气体脱保护得到产物1,总收率为85%(Scheme 11)。该合成方法先环合后脱保护,更易从反应液中提取出产物,大大提高收率。
2016年,胡海威等[30]报道以19为起始原料,经苄基保护、NaBH4还原、环合、脱苄基保护等一系列反应得到目标化合物1,收率为75%(Scheme 12)。该路线通过苄基对羟基进行保护,生成的 (S)-4-氯-3-苄氧基丁酸乙酯很容易从反应液中分离出来,而且苄基的脱保护反应比较彻底,因此整个制备方法的总收率较高。
1.3 1-丁烯-4-醇(27)为原料
2012年,Sudalai等[31]报道以27为原料,先用对甲苯磺酰氯对羟基进行保护得到3-丁烯基-4-甲基苯磺酸酯(28),再经间氯过氧苯甲酸(m-CPBA)环氧化得到2-(环氧乙烷-2-基)乙基-4-甲基苯磺酸酯(29),再环合得到产物1,收率为48%,ee值为98%,副产物 (R)-2-(环氧乙烷-2-基)乙基-4-甲基苯磺酸酯(30)的收率为47%。此外,他们还报道由27经Sharpless环氧化反应直接一步得到1,收率为95%,ee值为89%,该路线最大的优点是步骤短,反应条件温和;不足之处是试剂成本较高,ee值偏低(Scheme 13)。

Scheme 15
Scheme 16

Scheme 17
1.4 (S)-肉碱(31)为原料
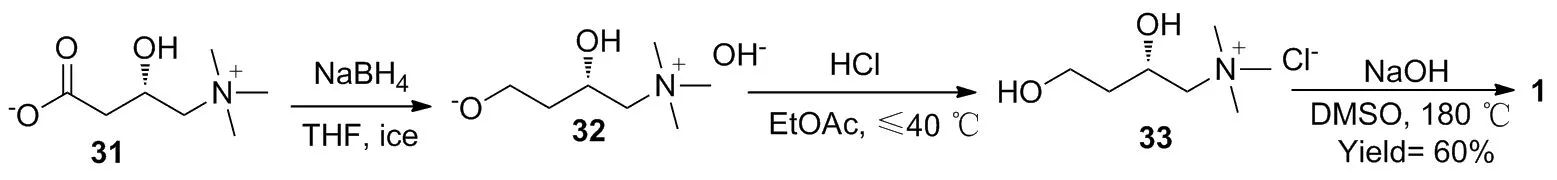
2015年,王平等[32]报道一种以31为起始原料,冰浴下经NaBH4还原得到 (S)-2,4-二羟基-N,N,N-三甲基丁胺碱(32),用盐酸成盐得到 (S)-2,4-二羟基-N,N,N-三甲基氯化丁胺(33),最后在碱性条件下高温关环得到1,总收率为60%,ee值99.3%(Scheme 14)。该路线原料价廉易得,反应步骤短,不足之处是最后一步使用了DMSO作溶剂,后处理比较麻烦。
不对称硼氢化反应可以合成手性仲醇类化合物。1986年,Brown等[33]采用手性硼催化剂2,3-二氢呋喃(34)实现了2,3-二氢呋喃(35)的不对称硼氢化还原,产物1的收率为92%,ee值为100%(Scheme 15)。该方法实现手性化合物1的高效合成,反应可以达到0.25 mol级别,具有潜在的工业化应用前景。
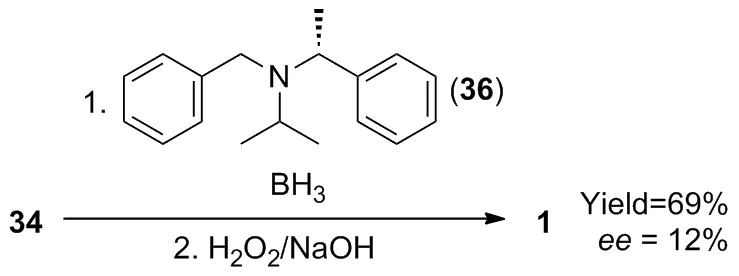
1987年,Periasamy等[34]利用手性路易斯碱 (R)-N-苄基-N-(1-苯基乙基)丙-2-胺(36)/BH3复合物实现34的不对称氢化,得到产物1的收率为69%,ee值仅12%(Scheme 16)。尽管该方法ee值较低,但为不对称还原合成 (S)-3-羟基四氢呋喃提供了一种新的催化体系。
1988年,Brown等[35]利用一种新的手性硼烷催化剂 [(1S,2R,3S,6R)-3,7,7-三甲基双环[4.1.0]庚-2-基]硼烷(37)实现2,5-二氢呋喃(38)的不对称还原,产物收率为53%,ee值为51%(Scheme 17)。该反应路线ee值较之前有较大提高,为1的不对称合成提供了借鉴。
1993年,Hayashi等[36]报道了不对称硅氢化合成1的方法。以38为起始原料,Cl3SiH作氢源,在手性配体(R)-(+)-MOP的催化下得到 (S)-3-三氯硅烷基四氢呋喃(40),再经Tamao等[11]报道的方法合成产物1,总收率为55%,ee值为95%(Scheme 18)。
2013年,Nobili等[37]报道采用酶解法获得1的方法。他们以3-羟基四氢呋喃(41)为原料,先用乙酰氯进行羟基保护,得到酯化产物3-乙酰氧基四氢呋喃(42),接着从水解酶中筛选出高对映选择性的碳酸酯酶(BsteE)[38-39],将其应用于42的不对称催化,得到产物1的收率为61%,ee值达100%(Scheme 19)。
2016年,胡海威等[40]以葡萄糖溶液为底物,解脂假丝酵母为发酵菌株,经扩大培养和发酵得到赤鲜醇粗品,经处理得到赤鲜醇溶液,将其作为原料,通入氢气,加入酸性溶液,搅拌反应,然后加入碱性溶液中和至中性使反应停止,最后萃取,减压蒸馏得到1,收率为95%,ee值为96%。该反应利用赤鲜醇在酸性环境下与氢气发生聚合反应制备(S)-3-羟基四氢呋喃,省去赤鲜醇干燥固化等多重工艺,简化步骤,制备方法简单有效,反应温和,有利于工业化生产。

Scheme 18

Scheme 19

Scheme 20
同年,Reetz等[41]报道以四氢呋喃-3-酮(43)为原料,在乙醇脱氢酶(ADH)催化下实现羰基的不对称还原,产物1的收率和ee值都较高(Scheme 20)。
目前,合成(S)-3-羟基四氢呋喃(1)的方法众多[42-45],各具优缺点。以L-苹果酸及其衍生物为原料的路线,原料价廉易得,反应条件温和,但由于中间体三醇水溶性较大难提纯而影响收率。1-丁烯-4-醇与(S)-肉碱作原料的研究相对较少。以2,3-二氢呋喃和2,5-二氢呋喃为原料的不对称合成1的方法较多,但由于催化剂难制备等原因,应用受到一定限制。直接对3-羟基四氢呋喃进行手性拆分成本较高,难度较大。相比而言,(S)-4-氯-3-羟基-丁酸乙酯为原料的合成路线步骤短,操作简便,收率较高,目前已实现工业化。除了化学合成法之外,生物发酵法也有可能在1的合成中发挥作用。相信在不久的将来,越来越多高效绿色经济的方法将被开发出来应用于1的工业合成。
[1] 孙万儒. 手性化合物的生物合成与转化[J].化工科技市场,2003,26(6):5-7.
[2] 徐雅妮,张凤宝,王燕. 手性化合物拆分方法的研究进展[J].天津化工,2002,6:20-22.
[3] 苟劲,刘永红,徐红. 不对称氢化反应技术在药物合成中的应用研究[J].重庆工学院学报,2006,20(5):136-142.
[4] 王彦美,韩杰,张良辅,等. 铂抗癌药物研究进展[J].合成化学,2003,11(1):27-31.
[5] 广常真治. 无脑回畸形治疗剂:CN 103282043[P].2013.[6] Wang X J, Zhang L, Weber D,etal. Efficient synthesis of empagliflozin, an inhibitor of SGLT-2, utilizingan AlCl3-promoted silane reduction of aβ-glycopyranoside[J].Org Lett,2014,16:4090-4093.
[7] 杜铁奇,郑亿,钟为慧,等. C-芳基糖苷类SGLT2抑制剂的合成研究进展[J].浙江化工,2015,46:19-23.[8] Michael R H, Roger D T, Chritopher T B,etal. Prodrugs of aspartyl protease inhibitors:US 6436989[P].2002.[9] 张娜,赵宁,虞心红,等. 安普那韦的合成[J].合成化学, 2008,16(1):115-117.
[10] 许学农. 阿法替尼的制备方法:CN 103242303[P].2013.[11] Wynberg H. The synthesis of 3-substituted furans[J].J Am Chem Soc,1958,80:364-366.
[12] Wynberg H, Bantjes A. 3-Hydroxytetrahydrofuran[J].Org Synth,1958,4:534.
[13] Tandon V K, Leusen A M, Wynberg H. Synthesis of enantiomericallypure (S)-(+)-3-hydroxytetrahydrofuranand its (R)-enantiomer from Malic or Tartaric Acid[J].J Org Chem,1983, 48(16):2767-2769.
[14] Hayashi H, Nakanishi K, Brandon C,etal. Structure and synthesis of dihydroxypentyluracil from bacteriophage SP-15 deoxyribonucleic acid[J].J Am Chem Soc,1973,95(24):8749-8757.
[15] Tang K C,Tropp B E, Engel R. The synthesis of phosphonic acid and phosphate analogs of glycerol-3-phosphate and related metabolites[J].Tetrahedron,1978,34:2873-2878.
[16] Hungerbiihler E, Seebach D, Wasmuth D. Doppelt und dreifach funktionalisierte, enantiomerenreine C4-synthesebausteine aus phydroxybuttersaure, apfelsaure und weinsaure[J].Helv Chim Acta,1981,64:1467-1487.[17] Honda Y, Katayama S, Izawa K,etal. New approaches to the industrial synthesis of HIV protease inhibitors[J].Org Biomol Chem,2004,2(14):2061-2070.[18] Bischofberger K, Bull J R. 14-Methyl steroids.part 3.synthesis of (±)14ɑ-methyl-9β-estradiol and related 14ɑ- methyl -19 -norsteroids[J].Tetrahedron,1985,41:365-374.
[19] 郑剑峰,苏贞夏,靳立人. 一种合成(S)-3-羟基四氢呋喃的方法:CN 1887880 [P].2007.
[20] Wynberg H, Staring E G J. Asymmetric synthesis of (S)-and (R)-Malic Acid from ketene and chloral[J].J Am Chem Soc,1982,104:166-168.
[21] 顾松林,汤玉亮,芮新生, 等. (S)-3-羟基-γ-丁内酯、(S)-3-羟基四氢呋喃联合生产方法:CN 101367780[P].2008.
[22] Yamaguchi A, Hiyoshi N, Shirai M,etal. Enhancement of cyclic ether formation from polyalcohol compound sin high temperature liquid water by high pressure carbon dioxide[J].Green Chem,2009,11(1):48-52.[23] Tomczyk K M, Guńka P A, Rokicki G,etal. Intramolecular etherification of five-membered cyclic carbonates bearing hydroxyalkyl groups[J].Green Chem,2012,14(6):1749-1758.
[24] 比克尔 M,黑克曼 G,马兰 C,等. 酮酯的氢化方法:CN 102844293[P].2012.
[25] Ribeiro G B, Ramos A D S, Fiaux S B,etal. Immobilized microorganisms in the reduction of ethyl 4-chloro acetoacetate[J].Tetrahedron:Asymmetry,2009,20:2263-2266.
[26] Srivastava G, Pal M, Jolly R S,etal. A highly efficient designer cell for enantioselective reduction of ketones[J].Catal Sci Technol,2015,5:105-108.
[27] He Y C, Zhang D P, Di J H,etal. Effective pretreatment of sugarcane bagasse with combination pretreatment and its hydrolyzates as reaction media for the biosynthesis of ethyl (S)-4-chloro-3-hydroxybutanoate by whole cells of E. coli CCZU-K14[J].Bioresource Technology,2016,211:720-726.
[28] 李勇智,宁斌科,丁秀丽. 手性化合物(S)-3-羟基四氢呋喃的合成[J].应用化工,2008,37:191-194.
[29] 陆军,顾铭,肖本良,等. 制备S-3-羟基四氢呋喃的新方法:CN 102477019[P].2010.
[30] 胡海威,丁靓,严辉,等. 一种(S)-3-羟基四氢呋喃的制备方法:CN 105669608[P].2016.
[31] Gadakh S K, Reddy R S, Sudalai A. Enantioselective synthesis of HIV protease inhibitor amprenavir via Co-catalyzed HKR of 2-(1-azido-2 phenylethyl)oxirane[J].Tetrahedron Asymmetry,2012,23:898-903.
[32] 王平,周文峰,张少平,等. (S)-3-羟基四氢呋喃和(R)-3-羟基四氢呋喃的制备方法:CN 104961711[P].2015.
[33] Brown H C, Vara Prasad J V N. Chiral synthesisviaorganoboranes.6.hydroboration.74.asymmetric hydroboration of representative heterocyclic olefins with diisopinocampheylborane.synthesis of heterocyclic boronates and heterocyclic alcohols of very high enantiomeric purity[J].J Am Chem Soc,1986,108(8):2049-2054. [34] Narayana C, Periasamy M. Hydroboration of pro-chiral olefins with chiral Lewis base-borane complexes: Relationship to the mechanism of hydroboration [J].J Chem Soc,1987,1857-1859.
[35] Brown H C, Vara Prasad J V N, Zaidlewicz M. Hydroboration. 82. Asymmetric hydroboration of representative cis-disubstituted and heterocyclic olefins with dicaranylboranes of high enantiomeric purity [J]J Org Chem,1988,53:2911-2916.[36] Uozumi Y, Hayashi T. Asymmetric hydroshylation of dihydrsfurans by use of palladium-MOP catalyst[J].Tetrahedron Lett,1993,34(14):2335-2338.
[37] Nobili A, Gall M G, Pavlidis I V. Use of ‘small but smart’ libraries to enhance the enantioselectivity of an esterase from Bacillus stearothermophilus towards tetrahydrofuran-3-ylacetate[J].FEBS Journal,2013,280(13):3084-3093.
[38] Burch J. The purification and properties of horse liver esterase[J].Bio Chem J,1954,58:415-426.
[39] Barker D L, Jencks W P. Pig liver esterase. Physical properties. Bio Chemistry,1969,8:879-3889.
[40] 胡海威,丁靓,严辉,等.一种基于赤鲜醇微生物制备(S)-3-经基四氢呋喃的方法:CN 105624227[P].2016.[41] Sun Z T, Lonsdale R, Reetz M T,etal. Catalytic asymmetric reduction of difficult-to-reduce ketones:Triple-code saturation mutagenesis of an alcohol dehydrogenase[J].ACS Catal,2016,6:1598-1605.
[42] Kakogawa K K, Takasago T M, Kakogawa Y Y,etal. Process for the preparation of 3-hydroxytetrahydrofuran:US 6359155[P].2000.
[43] Quan L G, Boo C J, Hong M H,etal. Process for the efficient preparation of 3-hydroxytetrahydrofuran:WO 2008093955[P].2008.
[44] 朱升,林文清,张晓梅. (S)-(+)-3-羟基四氢呋喃的合成工艺改进[J].合成化学,2008,16(5):609-610.[45] 王晓琴,朱德荣,何明华. 药物中间体(S)-3-羟基四氢呋喃的合成研究[J].化学研究与应用,2009,21:1070-1072.
Research Progress on Synthesis of (S)-3-Hydroxytetrahydrofuran
ZHENG Yi, WANG Hui, QI Liang, LING Fei, ZHONG Wei-hui*
(Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, College of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310014, China)
Preparation technology of (S)-3-hydroxytetrahydrofuran were reviewed with 45 references. Three commonly used synthetic methods were introduced in detail: (1) chiral substrates as the chiral source. (2) asymmetric synthesis using chiral catalysts. (3) asymmetric synthesisviaenzyme catalysis. The advantages and disadvantages of different methods were analyzed. The prospect of preparation process of 1 is presented.
(S)-3-hydroxytetrahydrofuran; synthesis; intermediate; review
2016-09-29;
2016-12-05
国家自然科学基金资助项目(21276238, 21676253)
郑亿(1991-),女,汉族,浙江衢州人,硕士研究生,主要从事手性药物及中间体的合成研究。 E-mail: 18767121220@163.com
钟为慧,教授,博士生导师, Tel. 0571-88320117, E-mail: weihuizhong@zjut.edu.cn
O626.11
A
10.15952/j.cnki.cjsc.1005-1511.2017.03.16248
猜你喜欢
化工设计(2022年4期)2023-01-02
贵州科学(2022年4期)2022-09-05
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
铜仁学院学报(2018年6期)2018-07-05
化工管理(2017年35期)2018-01-10
中成药(2017年5期)2017-06-13
中国洗涤用品工业(2016年2期)2016-02-28
中国塑料(2015年2期)2015-10-14
股市动态分析(2015年12期)2015-09-10
