
合成SGLT2抑制剂埃格列净的新方法
2017-04-01 02:52贺礼东吴启月刘亦斌顾洪伟陈友喜
合成化学 2017年3期
贺礼东, 吴启月, 刘亦斌, 顾洪伟, 何 姜, 陈友喜
(上海阳帆医药科技有限公司,上海 201203)
·制药技术·
合成SGLT2抑制剂埃格列净的新方法
贺礼东, 吴启月, 刘亦斌, 顾洪伟, 何 姜*, 陈友喜*
(上海阳帆医药科技有限公司,上海 201203)
以D-(+)-葡萄糖酸内酯为原料,经三甲硅基保护羟基后与5-溴-2-氯-4′-乙氧基二苯甲烷偶联制得(2S,3R,4S,5S,6R)-2-[4-氯-3-(4-乙氧苄基)苯基]-6-(羟甲基)-2-甲氧基四氢-2H-吡喃-3,4,5-三醇(2); 2经羟基保护、氧化和羟醛缩合等5步反应制得(3S,4S,5R,6S)-3,4,5-三(苄氧基)-6-[4-氯-3-(4-乙氧苄基)苯基]-2-(羟甲基)-6-甲氧基四氢-2H-吡喃-2-甲醛(7); 7经还原、脱苄同时关环制得埃格列净(1S,2S,3S,4R,5S)-5-[4-氯-3-(4-乙氧苄基)苯基]-1-(羟甲基)-6,8-二氧杂二环[3.2.1]辛烷-2,3,4-三醇,其结构经1H NMR和LC-MS表征。
D-(+)-葡萄糖酸内酯; 埃格列净; SGLT2抑制剂; 药物合成; 关环; 脱苄
钠-葡萄糖共转运体(SGLT)是一类葡萄糖转运蛋白,分为SCLT1和SGLT2[1]两种,在肾脏中均有表达,SGLTl分布较少,主要在近小管末端S3节段表达,完成肾滤液中10%的葡萄糖重吸收,而SGLT2主要分布在近小管Sl节段,负责肾滤液中90%的葡萄糖重吸收。所以,SGLT2的抑制剂可以通过阻断近曲小管对葡萄糖的重吸收而降低体内血糖浓度,从而达到治疗糖尿病的目的。各大制药公司以SGLT2为靶点合成了多个抑制剂。其中,辉瑞公司研发的(1S,2S,3S,4R,5S)-5-[4-氯-3-(4-乙氧苄基)苯基]-1-(羟甲基)-6,8-二氧杂二环[3.2.1]辛烷-2,3,4-三醇(10, Ertugliflozin,埃格列净)对SGLT2的抑制活性、SGLT1/SGLT2选择性都有优异的表现[2]。目前已报道多种合成10的方法[3-5]。Mascitti等[3]采用极性反转的方法将芳环与开链的糖片段连接,使用1,3-丙二硫醇会产生恶臭。Bernhardson等[4]步骤较短,但羟醛缩合与坎尼扎罗反应一锅同时进行,副产物较多。Bowles等[5]步骤较多且要用到格氏反应条件苛刻、难于操作。

Scheme 1
本文以D-(+)-葡萄糖酸内酯为原料,经三甲硅基保护羟基后[7]与5-溴-2-氯-4′-乙氧基二苯甲烷偶联制得(2S,3R,4S,5S,6R)-2-[4-氯-3-(4-乙氧苄基)苯基]-6-(羟甲基)-2-甲氧基四氢-2H-吡喃-3,4,5-三醇(2)[8-9]; 2经羟基保护、氧化和羟醛缩合等5步反应制得(3S,4S,5R,6S)-3,4,5-三(苄氧基)-6-[4-氯-3-(4-乙氧苄基)苯基]-2-(羟甲基)-6-甲氧基四氢-2H-吡喃-2-甲醛(7); 7经还原、脱苄同时关环制得10,其结构经1H NMR和LC-MS表征。
1 实验部分
1.1 仪器与试剂
Varian Mercury 400型核磁共振仪(CDCl3为溶剂,TMS为内标)。
干燥的THF用二苯甲酮作指示剂从金属钠中蒸馏获得;硅胶,200~300目;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1) (3R,4S,5R,6R)-3,4,5-三[(三甲硅基)氧]-6{[(三甲硅基)氧]甲基}四氢-2H-吡喃-2-酮(1)的合成
在反应瓶中加入葡糖酸内酯3.0 g(17 mmol)和无水四氢呋喃30 mL,于0~-10 ℃加入N-甲基吗啉13 mL(100 mmol)和三甲基氯硅烷15 mL(136 mmol),加毕,自然升至室温反应18 h。加入冰水50 mL,用乙酸乙酯(3×20 mL)萃取,合并萃取液,依次用2%磷酸二氢钠溶液和饱和食盐水洗涤有机相用硫酸钠干燥,减压蒸除溶剂得淡黄色液体1 8.0 g,收率100 %;1H NMRδ: 4.16(dt,J=7.6 Hz, 2.4 Hz, 1H), 3.98(d,J=7.9 Hz, 1H), 3.89(t,J=7.9 Hz, 1H), 3.82~3.71(m, 3H), 0.18(s, 9H), 0.16(s, 9H), 0.15(s, 9H), 0.11(s, 9H); MS(EI)m/z: 466[M+]。
(2) 2的合成
在反应瓶中加入5-溴-2-氯-4′-乙氧基二苯甲烷32.0 g(0.1 mol)和无水四氢呋喃400 mL,氩气保护,于-70 ℃注入正丁基锂41 mL(0.103 mol),加毕(控制温度-70~-80 ℃),保温反应20 min;注入1 50.0 g(0.11 mol),加毕,保温反应35 min;用注射器加入含甲烷磺酸7.8 mL(0.12 mol)的甲醇(20 mL)溶液,加毕,缓慢升至室温,反应5 h。加入饱和碳酸氢钠溶液500 mL,用二氯甲烷(3×150 mL)萃取,有机相用饱和食盐水洗涤,经硅胶柱层析[洗脱剂:A=V(二氯甲烷)/V(甲醇)=25/1]纯化得淡黄色无定形粉末2 13.0 g,收率29%;1H NMRδ: 7.52(d,J=2.0 Hz, 1H), 7.43(dd,J=8.4 Hz, 2.0 Hz, 1H), 7.33(d,J=8.4 Hz, 1H), 7.06(d,J=8.6 Hz, 2H), 6.77(d,J=8.6 Hz, 2H), 4.06(d,J=15.1 Hz, 1H), 3.96(m, 3H), 3.90(dd,J=12.2 Hz, 2.2 Hz, 1H), 3.79(dd,J=12.2 Hz, 5.4 Hz, 1H), 3.73(t,J=9.4 Hz, 1H), 3.56(ddd,J=9.4 Hz, 5.4 Hz, 2.2 Hz, 1H), 3.40(t,J=9.4 Hz, 1H), 3.07(d,J=9.4 Hz, 1H), 3.04(s, 3H), 1.33(t,J=7.0 Hz, 3H); LC-MSm/z: 406.90{[M-MeO-]+}。
(3) (2S,3R,4S,5S,6R)-6-{[(叔丁基二苯基硅基)氧]甲基}-2-[4-氯-3-(4-乙氧苄基)苯基]-2-甲氧基四氢-2H-吡喃-3,4,5-三醇(3)的合成
在反应瓶中加入2 7.462 g(17 mmol)和二氯甲烷50 mL,于0 ℃加入对二甲氨基吡啶(DMAP) 312 mg(2.55 mmol)、咪唑7.0 g(102 mmol)和叔丁基二苯基氯硅烷10.6 mL(40 mmol),加毕,于室温反应30 h。加入水100 mL,用二氯甲烷(3×50 mL)萃取,有机相用饱和食盐水洗涤,无水硫酸钠干燥,经硅胶柱层析(洗脱剂:A=30/1)纯化得淡黄色泡沫固体3 6.866 g, 收率60%;1H NMRδ: 7.75(dt,J=8.1 Hz, 1.6 Hz, 4H), 7.54(d,J=2.0 Hz, 1H), 7.42~7.30(m, 8H), 7.01(d,J=8.7 Hz, 2H), 6.73(d,J=8.7 Hz, 2H), 4.10~3.90(m, 7H), 3.76(t,J=9.1 Hz, 1H), 3.70(ddd,J=10.1 Hz, 5.1 Hz, 1.9 Hz, 1H), 3.57~3.51(m, 1H), 3.10(s, 3H), 1.33(t,J=7.0 Hz, 3H), 1.02(s, 9H); LC-MSm/z: 645.00 {[M-MeO-]+}。
(4) 叔丁基二苯基{(2R,3R,4S,5R,6S)-3,4,5-三(苄氧基)-6-[4-氯-3-(4-乙氧苄基)苯基]-6-甲氧基四氢-2H-吡喃-2-基}甲氧基硅烷(4)的合成
在反应瓶中加入3 6.788 g(10 mmol)和N,N-二甲基甲酰胺30 mL,于0 ℃加入60%氢化钠2 g(0.05 mol),加毕,于室温反应36 min;于0 ℃下缓慢滴加溴化苄6 mL(50 mmol),滴毕,自然升至室温,反应3 h。反应液倒入冰水100 mL中,乙酸乙酯(3×40 mL)萃取,有机相用饱和食盐水洗涤,用无水硫酸钠干燥,减压蒸除溶剂得淡黄色黏稠液体4 12 g,直接投入下步反应。
(5){(2R,3R,4S,5R,6S)-3,4,5-三(苄氧基)-6-[4氯-3-(4-乙氧苄基)苯基]-6-甲氧基四氢-2H-吡喃-2-基}甲醇(5)的合成
在反应瓶中加入4 12.0 g(10 mmol)、四氢呋喃50 mL和三水合四丁基氟化胺9.4 g(30 mmol),于室温反应20 h。加水100 mL,用乙酸乙酯(3×50 mL)萃取,萃取液依次用饱和食盐水洗涤,硫酸钠干燥,减压蒸除溶剂,经硅胶柱层析[洗脱剂:B=V(石油醚)/V(乙酸乙酯)=10/1]纯化得淡黄色黏稠液体5 6.0 g,两步收率83%;1H NMRδ: 7.37~7.27(m, 13H), 7.24~7.16(m, 3H), 7.03(d,J=8.6 Hz, 2H), 6.99~6.98(m, 2H), 6.76(d,J=8.6 Hz, 2H), 4.94~4.87(m, 3H), 4.69(d,J=10.9 Hz, 1H), 4.48(d,J=10.7 Hz, 1H), 4.19~4.14(m, 1H), 4.10~4.06(m, 1H), 3.97(q,J=7.0 Hz, 2H), 3.92~3.87(m, 3H), 3.79(dd,J=11.7 Hz, 4.0 Hz, 1H), 3.75~3.63(m, 2H), 3.30(d,J=9.6 Hz, 1H), 3.07(s, 3H), 1.39(t,J=7.0 Hz, 3H); LC-MSm/z: 675.90 {[M-MeO-]+}。
(6) (2S,3S,4S,5R,6S)-3,4,5-三(苄氧基)-6-[4-氯-3-(4-乙氧苄基)苯基]-6-甲氧基四氢-2H-吡喃-2-甲醛(6)的合成
在反应瓶中加入5 102 mg(0.144 mmol)和二氯甲烷2 mL,加入戴斯-马丁氧化剂122 mg(0.288 mmol),加毕,于室温反应1 h。加入硫代硫酸钠溶液10 mL,用二氯甲烷(3×5 mL)萃取,碳酸氢钠溶液洗涤,硫酸钠干燥,减压蒸除溶剂得淡黄色胶体6 167 mg,粗品直接投入下步反应。
(7) 7的合成
在反应瓶中加入6 167 mg(0.144 mmol)和二氧六环2 mL,加入37%的甲醛溶液1.44 mL(14.4 mmol),加毕,升温至70 ℃,加入氢氧化钠17 mg(0.432 mmol),加毕,反应17 h。冷却至室温,加入食盐水10 mL,用乙酸乙酯(2×5 mL)萃取,萃取液用硫酸钠干燥,减压蒸除溶剂得无色胶状固体7 147 mg,粗品直接投入下步反应。
(8) {(3S,4S,5R,6S)-3,4,5-三(苄氧基)-6-[4-氯-3-(4-乙氧苄基)苯基]-6-甲氧基四氢-2H-吡喃-2,2-二基}二甲醇(8)的合成
在反应瓶中加入7 147 mg(0.144 mmol)、四氢呋喃1 mL、甲醇2 mL和硼氢化钠11 mg(0.288 mmol),于室温反应30 min。加入食盐水10 mL,用乙酸乙酯(3×5 mL)萃取,萃取液用硫酸钠干燥,减压蒸除溶剂,经硅胶柱层析(洗脱剂:B=5/1)纯化得无色胶状固体8 38 mg,三步收率36%;1H NMRδ: 7.37~7.20(m, 16H), 7.07~7.04(m, 2H), 7.02(d,J=8.6 Hz, 2H), 6.79(d,J=8.6 Hz, 2H), 5.01~4.88(m, 3H), 4.68(d,J=10.8 Hz, 1H), 4.60(d,J=10.5 Hz, 1H), 4.39(t,J=9.8 Hz, 1H), 4.33(dd,J=11.7 Hz, 1.9 Hz, 1H), 4.08(d,J=15.7 Hz, 1H), 4.03~3.93(m, 4H), 3.90~3.80(m, 3H), 3.6(t,J=11.4 Hz, 1H), 3.25(d,J=9.9 Hz, 1H), 3.06(s, 3H), 2.95(dd,J=10.9 Hz, 1.9 Hz, 1H), 1.73(t,J=6.7 Hz, 1H), 1.40(t,J=7.0 Hz, 3H); LC-MSm/z: 706.95{[M-MeO-]+}。
(9) 10的合成
在反应瓶中加入8 80 mg(0.11 mmol)、四氢呋喃1 mL和甲醇1 mL,加入邻二氯苯0.12 mL(1.1 mmol) 和10%钯/碳68 mg(0.6 mmol),加氢气球,抽排空气三次,于室温反应18 h。过滤,滤液减压蒸除溶剂得黄色黏稠胶状液体10 30 mg,收率88%;1H NMRδ: 7.44(d,J=1.6 Hz, 1H), 7.38(dd,J=8.3 Hz, 1.6 Hz, 1H), 7.34(d,J=8.3 Hz, 1H), 7.07(d,J=8.6 Hz, 2H), 6.78(d,J=8.7 Hz, 2H), 4.13(d,J=7.5 Hz, 1H), 4.02(s, 2H), 3.97(q,J=7.0 Hz, 2H), 3.83(d,J=12.5 Hz, 1H), 3.77(d,J=8.4 Hz, 1H), 3.67(d,J=12.4 Hz, 1H), 3.64(t,J=8.0 Hz, 1H), 3.58(d,J=7.2 Hz, 1H), 3.54(d,J=7.5 Hz, 1H), 1.34(t,J=7.0 Hz, 3H); LC-MSm/z: 436.90 {[M+H]+}, 458.90{[M+Na]+}。
文献[6]报道先经酸关环后再脱苄制得目标产物。我们尝试对8进行脱苄反应制备9(Scheme 2),但所得产物为10(Scheme 1)。TLC显示Rf值与埃格列净标准对照样一致,LC-MSm/z: 436.90{[M+H]+}, 458.90 {[M+Na]+}为埃格列净准分子离子峰,氢谱数据与文献值[2]一致,其中δ3.75和3.57处dd峰分别为糖环2-H和4-H吸收峰,因1-位与5-位形成氧桥环后吡喃糖六元环发生的微小构型变化使2-位H与4-位H有向平伏键转变的趋势,故产生微小的“W”型远程耦合,本文氢谱未测得此耦合在误差范围内。因此由8可脱苄和关环同时进行一步制得10,路线更为简洁。
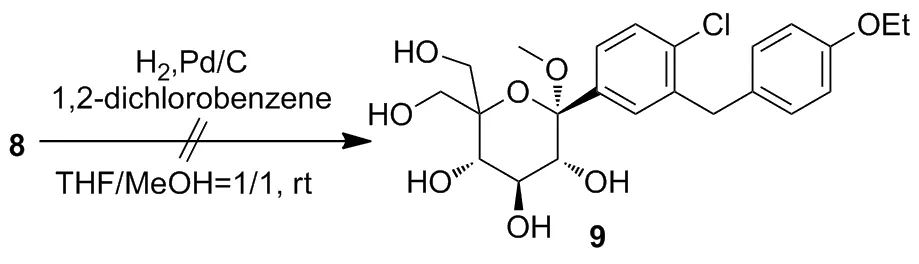
Scheme 2
以D-(+)-葡萄糖酸内酯为原料,经反应制得(1S,2S,3S,4R,5S)-5-[4-氯-3-(4-乙氧苄基)苯基]-1-(羟甲基)-6,8-二氧杂二环[3.2.1]辛烷-2,3,4-三醇,使合成路线更为简洁。本文对这一路线只做了初步探索,尚不适合工业化生产,可作为进一步优化的参考。
[1] Kanai Y, Lee W S, You G,etal. The human kidney low affinity Na+/glucose cotransporter SGLT2.Delineationof the major renal reabsorptive mechanism for D-glucose[J].J Clin Invest,1994,93(1):397-404.
[2] MascittiV, Maurer T S, Robinson R P,etal. Discovery of a clinical candidate from the structurally unique dioxa-bicyclo[3.2.1]octane class of sodium-dependent glucose cotransporter 2 inhibitors[J].J Med Chem,2011,54(8):2952-2960.
[3] Mascitti V, Préville C. Stereoselective synthesis of a dioxa-bicyclo[3.2.1]octane SGLT2 inhibitor[J].Org Lett,2010,12(13):2940-2943.
[4] Bernhardson D, Brandt T A, Hulford C A,etal. Development of an early-phase bulk enabling route to sodium-dependent glucose cotransporter 2 inhibitor ertugliflozin[J].Org Process Res Dev,2014,18(1):57-65.
[5] Bowles P, Brenek S J, Caron S,etal. Commercial route research and development for SGLT2 inhibitor candidate ertugliflozin[J].Org Process Res Dev,2014,18(1):66-81.
[6] 杨方龙,邓炳初,董庆,等. C-芳基葡萄糖苷衍生物,其制备方法及其在医药上的应用:WO 2012/019496 A1[P].2012.
[7] 赵文静,于秀玲,邵华,等. 含反式环己烷结构的C-葡萄糖苷类SGLT2抑制剂的合成及其降血糖活性Ⅱ[J].合成化学,2012,20(5):527-536.
[8] MengW, Ellsworth B A, Nirschl A A,etal. Discovery of dapagliflozin:A potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes[J].J Med Chem,2008,51(5):1145-1149.
[9] 邵华,赵桂龙,刘巍,等. SGLT2抑制剂Dapagliflozin的全合成[J].合成化学,2010,18(3):389-392.
〗
《合成化学》入网超星公司学术期刊“域出版”
为进一步方便读者,紧跟“互联网+”潮流,《合成化学》于2015年6月起正式加入超星公司学术期刊“域出版”。“域出版”是借助移动出版技术,通过“以智带栏”的模式,主要依托移动智能终端的在线学术交流互动平台。该平台的APP(支持Android和ios)已于7月底上线,欢迎广大读者下载使用。
《合成化学》编辑部
A Novel Method for Synthesis of SGLT2 Inhibitor Ertugliflozin
HE Li-dong, WU Qi-yue, LIU Yi-bin, GU Hong-wei, HE Jiang*, CHEN You-xi*
(Shanghai Sun-Sail Pharmaceutical Science & Technology Co., Ltd, Shanghai 201203, China)
(2S,3R,4S,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol(2) was prepared by coupling of 5-bromo-2-chloride-4′-ethoxydiphenylmethane with TMS-gluconolactone which D-(+)-gluconic acid lactone hydroxyl groups were protected by trimethylsilyl groups. (3S,4S,5R,6S)-3,4,5-tris(benzyloxy)-6-[4-chloro-3-(4-ethoxybenzyl)phenyl]-2-(hydroxymethyl)-6-methoxytetrahydro-2H-pyran-2-carbaldehyde(7) was prepared by a five-step reaction of hydroxyl protection, oxidation and aldol condensationetalfrom 2. Ertugliflozin, (1S,2S,3S,4R,5S)-5-[4-chloro-3-(4-ethoxybenzyl)phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol was prepared by formaldehyde reduction and debenzylation and cyclization from 7. The structure was confirmed by1H NMR and LC-MS.
D-(+)-gluconic acid lactone; Ertugliflozin; SGLT2 inhibitor; drug synthesis; cyclization; debenzylation
2016-08-20;
2017-01-03
贺礼东(1981-),男,汉族,山西应县人,硕士,主要从事药物合成研究。 E-mail: helidong2004@126.com
陈友喜,中级工程师, E-mail: cyxlzu@sina.com.cn; 何姜,主治医师, E-mail: hej_yd@sina.com
O621.3; R914.5
A
10.15952/j.cnki.cjsc.1005-1511.2017.03.16211
猜你喜欢
化学工程师(2022年5期)2022-05-11
山东化工(2020年15期)2020-09-01
铜仁学院学报(2018年6期)2018-07-05
中国洗涤用品工业(2016年2期)2016-02-28
烟草科技(2015年8期)2015-12-20
中国塑料(2015年2期)2015-10-14
云南中医学院学报(2015年2期)2015-07-31
中国洗涤用品工业(2015年9期)2015-02-28
世界热带农业信息(2014年10期)2014-11-13
中国塑料(2014年10期)2014-10-17
